
Hemocromatosis hereditaria para niños
Datos para niños Hemocromatosis hereditaria |
||
|---|---|---|
 Preparación teñida con azul de Prusia para revelar depósitos de hierro; muestra un patrón típico de acumulación de hierro de la hemocromatosis genética en heterocigosis (véase coloración azulada al ampliar la imagen).
|
||
| Especialidad | endocrinología hepatología |
|
| Sinónimos | ||
|
||
La hemocromatosis hereditaria (del griego haima: sangre y chróma: color) es una enfermedad hereditaria que afecta cómo el cuerpo maneja el hierro. Esta condición hace que se acumule demasiado hierro en los órganos y sistemas del cuerpo.
Es importante no confundirla con la hemosiderosis. La hemosiderosis es cuando hay mucho hierro en los tejidos, pero sin causar daño. Cuando el exceso de hierro sí causa problemas en órganos como el hígado, el páncreas o el corazón, entonces hablamos de hemocromatosis.
El hierro es vital para el cuerpo en cantidades normales. Ayuda en muchas funciones importantes. Sin embargo, si hay demasiado hierro, puede volverse dañino. El exceso de hierro puede generar sustancias que dañan los órganos.
Contenido
- ¿Cómo se descubrió la hemocromatosis?
- ¿Qué tan común es la hemocromatosis?
- Tipos de hemocromatosis
- ¿Cómo afecta el hierro a los órganos?
- Síntomas de la hemocromatosis
- ¿Cómo se diagnostica la hemocromatosis?
- Tratamiento de la hemocromatosis
- Pronóstico de la enfermedad
- Galería de imágenes
- Véase también
¿Cómo se descubrió la hemocromatosis?
La hemocromatosis fue descubierta por Armand Trousseau en 1865. Él describió un conjunto de síntomas que incluían diabetes, piel más oscura y problemas de hígado. En 1871, Trousseau llamó a esta combinación "diabetes bronceada".
En 1889, von Recklinghausen le dio el nombre de "hemocromatosis". Él descubrió que el hierro era el metal que se acumulaba en los tejidos. También fue el primero en sospechar que era una enfermedad hereditaria.
En 1935, Joseph H. Sheldon confirmó que era una enfermedad genética. Se dio cuenta de que afectaba cómo el cuerpo usa el hierro.
Un gran avance ocurrió en 1975. Simon y sus colegas descubrieron que la enfermedad estaba relacionada con un gen específico. Este gen se encuentra en el cromosoma 6.
En 1996, John N. Feder y su equipo identificaron el gen HFE. También encontraron las dos mutaciones principales asociadas a la enfermedad. Estas mutaciones son C282Y y H63D.
En el año 2000, se descubrió que algunos pacientes no tenían estas mutaciones. Esto llevó a identificar otros genes implicados en los años siguientes. A partir de 2004, se actualizó la forma de clasificar la hemocromatosis.

¿Qué tan común es la hemocromatosis?
La hemocromatosis hereditaria es la enfermedad genética más común en los países occidentales. Afecta a 1 de cada 200 personas. Los estudios muestran que es más común en personas de ascendencia europea, especialmente celta. La migración de los pueblos celtas podría explicar cómo se extendió la enfermedad.
También hay diferencias entre hombres y mujeres. Los hombres la padecen con más frecuencia que las mujeres, en una proporción de 3 a 1. Esto se debe a que los cambios naturales en el cuerpo de las mujeres, como la menstruación, ayudan a que pierdan hierro. Esto impide que se acumule en grandes cantidades.
Tipos de hemocromatosis
La enfermedad puede presentarse de dos formas:
- Hemocromatosis hereditaria o primaria: Se debe a un cambio en los genes.
- Hemocromatosis adquirida o secundaria: Es causada por otras enfermedades o situaciones.
Hemocromatosis hereditaria: causas genéticas

La hemocromatosis hereditaria es causada por cambios en el gen HFE. Este gen se encuentra en el cromosoma 6. El gen HFE produce una proteína que ayuda a regular la absorción de hierro. Se encuentra en órganos como el hígado y el intestino delgado.
Las mutaciones más comunes son:
- Mutación C282Y: Un cambio en el ADN que altera la proteína HFE. Del 85 al 100% de los pacientes tienen esta mutación.
- Mutación H63D: Otro cambio en el ADN, menos frecuente.
Esta enfermedad se hereda de forma autosómica recesiva. Esto significa que para padecerla, una persona debe heredar una copia del gen mutado de cada uno de sus padres. Si solo hereda una copia, es "portador" y puede transmitir la enfermedad, pero no la sufre.
No todas las personas con las mutaciones desarrollan la enfermedad grave. Sin embargo, sí tienen un mayor riesgo de acumular hierro. Otros factores pueden influir, como la cantidad de hierro en la dieta o ciertas infecciones.
Existen otros tipos de hemocromatosis primaria, causadas por mutaciones en otros genes:
| Tipo | OMIM | Gen mutado | Descripción |
|---|---|---|---|
| Hemocromatosis tipo 1 o hemocromatosis hereditaria (HH) |
|
HFE: gen que codifica para la proteína que lleva su nombre, reguladora de la absorción de hierro | Aumentan los niveles de hierro libre y su posterior absorción intestinal |
| Hemocromatosis tipo 2A o hemocromatosis juvenil |
|
HJV: codifica para la hemojuvelina, proteína moduladora de la hepcidina | Depósito de hierro en los tejidos antes de los 30 años en ambos sexos |
| Hemocromatosis tipo 2B o hemocromatosis juvenil |
|
HAMP: gen que codifica para la proteína hepcidina | El déficit de HAMP provoca sobrecarga de hierro |
| Hemocromatosis tipo 3 |
|
TfR2: codifica para el receptor 2 de la transferrina | Clínica indistinguible de la hemocromatosis tipo 1 |
| Hemocromatosis tipo 4 |
|
SLC40A1: gen que codifica para la ferroportina | Herencia autosómica dominante. Clínica similar a hemocromatosis tipo 1 |
Hemocromatosis tipo 3
El receptor de la transferrina 2 (TfR2) es una proteína que ayuda a que el hierro entre en las células. Se encuentra principalmente en el hígado y el intestino. Los síntomas de este tipo son muy parecidos a los de la hemocromatosis tipo 1, pero suelen aparecer a edades más tempranas.
Hemocromatosis adquirida: otras causas
La hemocromatosis secundaria ocurre cuando el cuerpo acumula hierro por otras razones, como:
- Múltiples transfusiones de sangre: Algunas enfermedades de la sangre requieren muchas transfusiones. Esto puede llevar a un exceso de hierro.
- Anemia hemolítica: Cuando los glóbulos rojos se destruyen, liberan hierro. Este hierro puede acumularse en los tejidos.
- Enfermedades del hígado: Como la hepatitis C.
- Consumo excesivo de hierro: Comer demasiados alimentos con mucho hierro puede saturar el sistema de absorción.
- Porfiria cutánea tarda: Una enfermedad de la piel que a menudo se asocia con acumulación de hierro en el hígado.
- Sobrecarga de hierro africana: Se cree que es causada por el consumo de bebidas fermentadas en recipientes de hierro.
¿Cómo afecta el hierro a los órganos?
El cuerpo humano tiene entre 4 y 5 gramos de hierro. La mayor parte está en la hemoglobina (en la sangre). Cada día se pierde una pequeña cantidad de hierro. En la hemocromatosis, el cuerpo absorbe mucho más hierro del que necesita.
Esto ocurre por dos mecanismos principales:
- Criptas intestinales: Normalmente, una proteína llamada HFE ayuda a los intestinos a saber cuánto hierro hay en la sangre. Si la HFE está mutada, no detecta bien el hierro. Esto hace que se absorba más hierro de lo necesario.
- Hepcidina: La hepcidina es una sustancia del hígado que regula la liberación de hierro a la sangre. En la hemocromatosis, hay una deficiencia de hepcidina. Esto provoca una sobrecarga de hierro sin control.
Síntomas de la hemocromatosis
Hay dos características comunes en todos los tipos de hemocromatosis: el exceso de hierro en todo el cuerpo y el daño a los órganos. El hígado es el órgano más afectado. Otros tejidos que sufren son el páncreas, el corazón, las glándulas endocrinas, las articulaciones y la piel.
Aunque el problema genético existe desde el nacimiento, los síntomas suelen aparecer en la edad adulta. Esto ocurre cuando el exceso de hierro es más grave. La evolución de la hemocromatosis se divide en tres etapas:
- Etapa I: No hay sobrecarga de hierro.
- Etapa II: Hay sobrecarga de hierro, pero sin síntomas graves. Los síntomas son muy generales, como cansancio o falta de energía.
- Etapa III: Hay sobrecarga de hierro y síntomas clínicos. Aparecen problemas en los órganos. Los síntomas más comunes son problemas de hígado, páncreas y piel. A menudo se le llama "diabetes bronceada".
Problemas en el hígado
.jpg)
El hígado es el órgano más afectado por el exceso de hierro. El hierro se deposita en las células del hígado. Esto puede causar fibrosis hepática y, si avanza, cirrosis. También aumenta el riesgo de desarrollar tumores malignos en el hígado. Un signo claro es el agrandamiento del hígado (hepatomegalia).
Problemas en el páncreas
Aproximadamente la mitad de las personas con hemocromatosis desarrollan diabetes. Esto puede deberse al exceso de hierro en las células del páncreas que producen insulina. También puede ser por resistencia a la insulina debido al daño hepático.
Daño al corazón
El exceso de hierro en las fibras musculares del corazón puede causar dos problemas:
- Insuficiencia cardíaca congestiva: El corazón no bombea sangre de manera eficiente. Esto causa cansancio y retención de líquidos.
- Arritmias: Alteraciones en el ritmo del corazón, como latidos muy lentos o muy rápidos.
El riesgo de muerte por problemas cardíacos es mucho mayor en estos pacientes.
Problemas hormonales
El exceso de hierro puede afectar el hipotálamo y la hipófisis. Esto puede causar:
- Hipogonadismo secundario: En hombres, puede llevar a problemas como la atrofia testicular y la disminución del deseo.
- Hipotiroidismo secundario: El hierro interfiere con la producción de hormonas tiroideas.
- Hipoparatiroidismo secundario: El exceso de hierro afecta la función de las glándulas paratiroides.
Problemas en las articulaciones
La afección de las articulaciones es común, afectando a un 30-40% de los pacientes. El hierro y otros depósitos dañan la membrana sinovial, el cartílago y el hueso. Esto causa una osteoartritis degenerativa, que es progresiva y crónica. Las articulaciones más afectadas son las de las manos, muñecas, rodillas, hombros y caderas.
Cambios en la piel
Aproximadamente el 70% de los pacientes tienen la piel más oscura (hiperpigmentación). A menudo se usa el término "diabetes bronceada" cuando la hemocromatosis presenta cirrosis, piel oscura y diabetes. También pueden observarse las uñas aplanadas y una disminución del vello corporal.
Otras manifestaciones
Los pacientes con hemocromatosis pueden tener otros síntomas menos comunes. Por ejemplo, problemas en los ganglios basales pueden causar movimientos involuntarios. También son más susceptibles a ciertas infecciones, causadas por microorganismos que crecen mejor con hierro.
¿Cómo se diagnostica la hemocromatosis?
A menudo, la hemocromatosis se detecta con análisis de sangre de rutina. Esto se debe a que los síntomas específicos suelen aparecer tarde. Para confirmar la enfermedad, se sigue un proceso: 1. Se evalúan los síntomas del paciente. 2. Se realizan análisis de sangre para confirmar la sospecha. 3. Si los análisis muestran alteraciones, se hace una prueba genética. 4. Finalmente, se puede hacer una biopsia de hígado para ver el daño.
Pruebas de sangre
Tres valores son muy útiles para el diagnóstico:
- Saturación de transferrina: Un valor superior al 45% en mujeres o 55% en hombres es anormal.
- Ferritina sérica: Cifras por encima de 350 ng/ml se consideran altas. Este valor indica la cantidad de hierro almacenado.
- Hierro sérico: Los niveles de hierro en sangre pueden variar mucho. Por eso, su uso como marcador principal es limitado.
Análisis genético

Si las pruebas de sangre muestran alteraciones, se realiza un Diagnóstico Molecular. Con una prueba de ADN se buscan las mutaciones C282Y y/o H63D. Esto confirma la enfermedad. Si el resultado es negativo para estas mutaciones, no se descarta la enfermedad. Se necesita un seguimiento y descartar otras causas. Este análisis es muy fiable, con más del 99% de precisión.
Biopsia de hígado

Es un método más invasivo. Permite saber la cantidad de hierro acumulado y el grado de daño en el hígado. También ayuda a descartar otras causas de daño hepático.
Se recomienda una biopsia si:
- Los pacientes tienen ferritina alta (más de 1000 ng/ml), transaminasas elevadas o agrandamiento del hígado.
- Los pacientes tienen pruebas genéticas negativas, pero niveles altos de saturación de transferrina y ferritina.
En la hemocromatosis primaria, el hierro se acumula en las células del hígado. En la secundaria, primero se acumula en otras células del hígado y luego en las células hepáticas.
Tratamiento de la hemocromatosis
El objetivo del tratamiento es reducir el exceso de hierro. La terapia varía según la causa de la enfermedad. Para la hemocromatosis primaria, el tratamiento principal es la flebotomía. Para la secundaria, se usan medicamentos llamados quelantes de hierro. Si hay complicaciones graves en el hígado, se puede considerar un trasplante. También es importante seguir una dieta especial y tomar otros medicamentos para los síntomas.
Flebotomías
Las flebotomías consisten en extraer sangre regularmente para disminuir el exceso de hierro. Al principio, se extraen 500 ml de sangre una vez a la semana durante 2 o 3 años. Esto reduce los niveles de hierro en el cuerpo.
Cuando los niveles de hierro bajan lo suficiente, se pasa a las flebotomías de mantenimiento. Estas se hacen con menos frecuencia, cada 3 o 4 meses. Este tratamiento suele ser de por vida.
Las flebotomías pueden reducir el tamaño del hígado, el cansancio y la debilidad. También ayudan a controlar la diabetes.
Quelantes de hierro

{kind=link}
Estos medicamentos se usan para las sobrecargas de hierro secundarias. Los quelantes se unen al hierro y lo hacen soluble. Así, evitan que el hierro cause daño. Hay tres quelantes principales:
- Deferoxamina: Se administra por inyección.
- Deferiprona: Se usa como alternativa o en combinación. Es más fácil de tomar, pero menos eficaz.
- Deferasirox: Se toma por vía oral una vez al día. Requiere supervisión médica por sus efectos secundarios.
Otros medicamentos
Muchos síntomas de la hemocromatosis no desaparecen solo con flebotomías o quelantes. Se necesitan otros medicamentos para controlarlos. Por ejemplo, se pueden usar antiinflamatorios para el dolor en las articulaciones. También se usan medicamentos para la diabetes y los problemas cardíacos.
Dieta especial
Es importante que los pacientes sigan una dieta adecuada:
- Evitar el consumo de alcohol, ya que puede dañar el hígado.
- Limitar el consumo de carne roja, que tiene mucho hierro.
- No consumir pescado ni mariscos crudos, por el riesgo de infecciones.
- Evitar suplementos vitamínicos que contengan hierro.
- Limitar la vitamina C, ya que puede aumentar la absorción de hierro.
Trasplante de hígado
Si hay una complicación grave en el hígado, como una insuficiencia hepática, se puede considerar un trasplante. El riesgo de muerte es mayor en estos pacientes debido a otros problemas asociados.
Pronóstico de la enfermedad
El pronóstico es mejor cuanto antes se diagnostique la enfermedad y se inicie el tratamiento. También influye el grado de daño en los tejidos.
En general, las personas con hemocromatosis tienen una esperanza de vida menor que el resto de la población. Las principales causas de muerte son la insuficiencia cardíaca, la insuficiencia hepática y el cáncer de hígado.
La calidad de vida del paciente también puede verse afectada. La cirrosis, la diabetes y, sobre todo, los problemas en las articulaciones influyen negativamente en su día a día.
Galería de imágenes
-
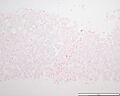
Preparación teñida con azul de Prusia para revelar depósitos de hierro; muestra un patrón típico de acumulación de hierro de la hemocromatosis genética en heterocigosis (véase coloración azulada al ampliar la imagen).
-

Armand Trousseau, descubrió la hemocromatosis.
-
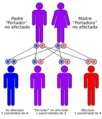
Ejemplo de herencia autosómica recesiva.
-
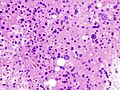
Imagen histológica de un hepatocarcinoma en un paciente con cirrosis hepática.
-

Taquicardia ventricular en un paciente con hemocromatosis (Derivación II del ECG).
-
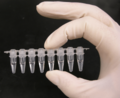
Tubos de PCR con muestra de ADN.
-
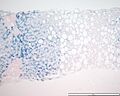
Acumulación de hierro de grado 3 en hepatocitos que muestra un patrón de distribución acinar consistente con una hemocromatosis genética homocigótica (preparado con tinción azul de Prusia de Perl).
-
Modelo molecular de la deferoxamina, un quelante del hierro.
Véase también
 En inglés: Hereditary haemochromatosis Facts for Kids
En inglés: Hereditary haemochromatosis Facts for Kids
