
Geoquímica para niños
La geoquímica es una ciencia que combina la química y la geología para entender cómo funcionan los sistemas de la Tierra, como la corteza terrestre y los océanos. También estudia el sistema solar completo, ayudando a comprender cómo se forman los planetas y cómo se mueven los materiales dentro de la Tierra.
Esta ciencia investiga la composición y el movimiento de los elementos químicos en nuestro planeta. Busca saber cuánto hay de cada elemento y cómo se distribuyen. También estudia cómo estos elementos se mueven entre las diferentes capas de la Tierra: la litósfera (rocas), la hidrósfera (agua), la atmósfera (aire) y la biósfera (seres vivos). Para ello, observa los cambios en las rocas y minerales.
Los elementos químicos más abundantes en la Tierra, de mayor a menor cantidad, son: oxígeno, silicio, aluminio, hierro, calcio, sodio, potasio y magnesio.
Contenido
- Historia de la Geoquímica
- Ramas de la Geoquímica
- Áreas de Investigación Geoquímica
- Elementos Químicos y la Tierra
- Diferenciación y Mezcla de Materiales
- Ciclos Geoquímicos
- Abundancia de Elementos en el Sistema Solar
- Composición de la Corteza Terrestre
- Metales Traza en el Océano
- Galería de imágenes
- Véase también
Historia de la Geoquímica

El término "geoquímica" fue usado por primera vez en 1838 por el químico suizo-alemán Christian Friedrich Schönbein. Él pensó que se necesitaba una "geoquímica comparativa" para entender el origen de los planetas y su materia.
Sin embargo, durante mucho tiempo, se usó más el término "geología química". Los geólogos y los químicos no trabajaban mucho juntos.
La geoquímica se convirtió en una ciencia separada cuando se crearon laboratorios importantes. Uno de los primeros fue el del Servicio Geológico de los Estados Unidos (USGS) en 1884. Allí, comenzaron a estudiar de forma organizada la química de las rocas y los minerales. Frank Wigglesworth Clarke, químico principal del USGS, notó que los elementos menos abundantes suelen tener pesos atómicos más altos.
Desde 1850, se empezó a investigar la composición de los meteoritos y a compararla con las rocas de la Tierra. En 1901, Oliver C. Farrington sugirió que, aunque hubiera diferencias, las cantidades relativas de los elementos deberían ser similares. Esto dio origen a la cosmoquímica, que nos ha enseñado mucho sobre cómo se formaron la Tierra y el sistema solar.
A principios del siglo XX, científicos como Max von Laue y William Lawrence Bragg descubrieron que los rayos X podían usarse para ver la estructura de los cristales. En las décadas de 1920 y 1930, Victor Goldschmidt aplicó estos métodos a muchos minerales comunes. Él estableció reglas sobre cómo se agrupan los elementos.
La geoquímica, como ciencia que explica la historia de los elementos en la Tierra, solo pudo desarrollarse cuando se entendió el concepto de elemento químico y la estructura de los átomos. Algunos logros importantes antes de que la geoquímica existiera formalmente incluyen:
- ~500 a.C.: Demócrito (griego) propuso la idea del átomo.
- 371-286 a.C.: Teofrasto (griego) estudió los minerales, rocas y suelos.
- Año 79: Plinio el Viejo (romano) recopiló datos sobre minerales.
- 1711-1765: Mijaíl Lomonósov (ruso) escribió un tratado que hablaba de la composición de las capas de la Tierra y el origen de los minerales, aunque no usó el nombre "geoquímica".
Ramas de la Geoquímica
La geoquímica tiene varias ramas especializadas:
- Geoquímica acuosa: Estudia cómo se comportan los elementos en el agua, como el cobre, el azufre y el mercurio, y cómo se mueven entre la atmósfera, la tierra y el agua.
- Biogeoquímica: Se enfoca en cómo los seres vivos afectan la química de la Tierra.
- Cosmoquímica: Analiza la distribución de los elementos y sus isótopos en el universo.
- Geoquímica de isótopos: Determina las cantidades de elementos y sus isótopos en la Tierra.
- Geoquímica orgánica: Estudia el papel de los procesos y compuestos que provienen de organismos vivos o antiguos.
- Fotogeoquímica: Investiga las reacciones químicas causadas por la luz en la superficie terrestre.
- Geoquímica regional: Se aplica a estudios ambientales, de agua y de búsqueda de minerales.
Áreas de Investigación Geoquímica
Los geoquímicos investigan en diversas áreas:
- Prospección geoquímica: Buscar minerales y recursos naturales.
- Geoquímica de reservorio: Estudiar la composición de depósitos de petróleo y gas.
- Geoquímica de elementos traza: Analizar elementos que están en muy pequeñas cantidades.
- Geoquímica de isótopos:
* Usar isótopos para determinar la edad de minerales y rocas (datación radiométrica). * Estudiar las variaciones de isótopos estables en materiales naturales.
- Geoquímica ambiental:
* Estudiar la distribución de elementos y compuestos dañinos en el ambiente. * Investigar la contaminación hídrica (del agua), del suelo y de la contaminación atmosférica (del aire). * Geoquímica forense: Aplicar principios geoquímicos para resolver crímenes o investigar eventos.
Elementos Químicos y la Tierra
Los elementos químicos son los componentes básicos de todo. Se identifican por su número atómico (Z), que es el número de protones en el núcleo atómico. Un elemento puede tener diferentes números de neutrones; a estas variaciones se les llama isótopos. Por ejemplo, el cloro tiene isótopos como el 35Cl y el 37Cl.
En geoquímica, los isótopos estables se usan para seguir rutas químicas, mientras que los isótopos inestables (radiactivos) se usan para datar muestras.
El comportamiento químico de un átomo depende de cómo están organizados sus electrones, especialmente los más externos. Esto se refleja en la tabla periódica. Los elementos se agrupan en categorías como metales alcalinos, metales de transición, halógenos y gases nobles.
Una forma útil de clasificar los elementos en geoquímica es la clasificación de Goldschmidt, que los divide en cuatro grupos:
- Litófilos: Se unen fácilmente con el oxígeno. Incluyen elementos como el sodio, potasio, silicio y aluminio. Son comunes en la corteza terrestre, formando silicatos.
- Siderófilos: Se unen fácilmente con el hierro. Incluyen hierro, cobalto y níquel. Tienden a concentrarse en el núcleo de la Tierra.
- Calcófilos: Se unen fácilmente con el azufre. Incluyen cobre, plata y zinc. Forman sulfuros.
- Atmófilos: Se combinan fácilmente con el oxígeno y el nitrógeno. Incluyen oxígeno, nitrógeno, hidrógeno y gases nobles. Dominan la atmósfera.
Dentro de cada grupo, algunos elementos son "refractarios" (estables a altas temperaturas) y otros son "volátiles" (se evaporan fácilmente).
Diferenciación y Mezcla de Materiales
La composición química de la Tierra y otros cuerpos celestes se forma por dos procesos opuestos: la diferenciación y la mezcla.
- Diferenciación: Es la separación de materiales en distintas regiones. Por ejemplo, en el manto de la Tierra, algunos materiales se funden y suben para formar basalto, mientras que otros más densos se quedan abajo. La erosión también diferencia el granito en arcilla, arenisca y minerales disueltos.
- Mezcla: Es la combinación de materiales. Después de que una placa oceánica se hunde en el manto, la convección (movimiento de calor) mezcla las partes. El metamorfismo (cambio de rocas por calor y presión) y la fusión de rocas pueden volver a mezclar elementos. En el océano, los organismos pueden causar diferenciación, pero su disolución y desechos mezclan los materiales de nuevo.
Fraccionamiento de Elementos
El fraccionamiento es una distribución desigual de elementos e isótopos. Puede ocurrir por reacciones químicas, cambios de estado (como de sólido a líquido), o por radioactividad.
A gran escala, la diferenciación planetaria es la separación física y química de un planeta en regiones distintas. Por ejemplo, los planetas rocosos como la Tierra tienen núcleos ricos en hierro y mantos y cortezas ricos en silicatos.
El fraccionamiento isotópico ocurre cuando los isótopos (átomos del mismo elemento con diferente número de neutrones) se separan. Los isótopos más pesados son más estables y tienden a concentrarse en las fases más densas o en compuestos con mayor oxidación.
Las proporciones de isótopos se comparan con un estándar. Por ejemplo, el azufre tiene isótopos como el 32S y el 34S. La proporción de sus concentraciones se expresa en partes por mil (‰) para mostrar las pequeñas diferencias.
Equilibrio y Cinética
- Fraccionamiento de equilibrio: Ocurre cuando sustancias o fases están en equilibrio. Las fases más pesadas prefieren los isótopos más pesados. Este fraccionamiento es mayor a temperaturas más bajas.
- Fraccionamiento cinético: Ocurre cuando no hay equilibrio. Los isótopos más ligeros suelen tener enlaces más débiles, reaccionan más rápido y enriquecen los productos de la reacción. El fraccionamiento biológico es un tipo de fraccionamiento cinético, donde los organismos prefieren los isótopos más ligeros porque requieren menos energía para romper sus enlaces.
Ciclos Geoquímicos
Los elementos químicos se mueven y cambian de concentración en lo que llamamos "ciclos geoquímicos". Para entender estos cambios, los geoquímicos dividen la Tierra en "depósitos geoquímicos" (como el océano o la atmósfera). En un "modelo de caja", cada depósito es una caja con entradas y salidas de elementos.
Estos modelos suelen incluir retroalimentación, lo que significa que la entrada o salida de un depósito depende de la concentración del elemento. Esto ayuda a mantener el sistema en un estado estable. El "tiempo de residencia" es el tiempo que un elemento permanece en un depósito.
Abundancia de Elementos en el Sistema Solar
Composición del Sistema Solar

La composición del sistema solar es similar a la de muchas otras estrellas. Se cree que se formó a partir de una nube de gas y polvo (nebulosa solar) con una composición uniforme. La fotosfera del Sol (su superficie visible) tiene una composición similar al resto del sistema solar.
Los dos elementos más abundantes por masa son el hidrógeno (74.9%) y el helio (23.8%). Todos los demás elementos juntos solo representan el 1.3%. En general, la abundancia de elementos disminuye a medida que aumenta su número atómico. Los elementos con número atómico par son más comunes que los impares (regla de Oddo-Harkins).
La abundancia de elementos se debe principalmente a dos factores: el hidrógeno, el helio y parte del litio se formaron poco después del Big Bang, mientras que el resto se creó dentro de las estrellas.
Meteoritos y su Importancia
Los meteoritos tienen diferentes composiciones. Las condritas son un tipo de meteorito que no ha cambiado mucho desde la formación del sistema solar, hace unos 4560 millones de años. Un tipo especial, la condrita CI, tiene una composición muy parecida a la del Sol, lo que las convierte en una excelente fuente para estudiar la composición del sistema solar temprano.
Planetas Gigantes

Los planetas del sistema solar se dividen en:
- Planetas terrestres: Mercurio, Venus, Tierra y Marte. Son pequeños y rocosos.
- Planetas gigantes: Dominados por hidrógeno y helio, con menor densidad. Se subdividen en:
* Gigantes gaseosos: Júpiter y Saturno. * Gigantes helados: Urano y Neptuno, con grandes núcleos de hielo.
La mayor parte de la información sobre la composición de los planetas gigantes proviene de la espectroscopia, que analiza la luz que emiten o reflejan. Así se detectaron elementos como hidrógeno, metano y amoniaco en Júpiter. Las sondas espaciales, como la sonda Galileo a Júpiter y la sonda Cassini a Saturno, han proporcionado datos más directos.
Los modelos actuales sugieren que los cuatro planetas gigantes tienen núcleos de roca y hielo de tamaño similar. Sin embargo, la proporción de hidrógeno y helio disminuye de Júpiter a Urano y Neptuno. Los gigantes gaseosos son principalmente hidrógeno y helio, mientras que los gigantes helados son principalmente elementos más pesados (oxígeno, carbono, nitrógeno, azufre) en forma de agua, metano y amoniaco.
Planetas Terrestres
Se cree que los planetas terrestres se formaron del mismo material que los gigantes, pero perdieron la mayoría de los elementos ligeros. Su composición se vio afectada por el enfriamiento de la nebulosa solar.
La información sobre Marte, Venus y Mercurio proviene de misiones espaciales que usaron espectrómetros de rayos gamma para medir la composición de sus cortezas. También se estudian meteoritos de Marte que han llegado a la Tierra.
Los modelos de formación planetaria sugieren que la composición de cada planeta terrestre dependió de la temperatura en su "zona de alimentación" en la nebulosa solar. Por ejemplo, Mercurio se formó a altas temperaturas, con mucho hierro metálico. La Tierra se formó a temperaturas más bajas, incorporando sulfuros de hierro y silicatos.
Composición de la Corteza Terrestre

Los componentes más comunes de las rocas son casi todos óxidos. El oxígeno constituye un poco más del 47% de la corteza terrestre, principalmente combinado en óxidos como la sílice, la alúmina y los óxidos de hierro. La sílice forma silicatos, que son los minerales más comunes en las rocas ígneas.
Según cálculos basados en análisis de rocas, la composición promedio de la corteza terrestre es:
- SiO2 (sílice) = 59.71%
- Al2O3 (alúmina) = 15.41%
- Fe2O3 (óxido de hierro) = 2.63%
- FeO (óxido ferroso) = 3.52%
- MgO (óxido de magnesio) = 4.36%
- CaO (óxido de calcio) = 4.90%
- Na2O (óxido de sodio) = 3.55%
- K2O (óxido de potasio) = 2.80%
- H2O (agua) = 1.52%
- TiO2 (dióxido de titanio) = 0.60%
- P2O5 (óxido de fósforo) = 0.22%
Todos los demás componentes están en cantidades muy pequeñas.
Estos óxidos se combinan para formar minerales. Por ejemplo, el óxido de potasio y el óxido de sodio se combinan para producir feldespatos. El ácido fosfórico con cal forma la apatita. El dióxido de titanio con óxido ferroso forma la ilmenita.
Minerales Comunes en la Corteza
La corteza terrestre está compuesta en un 90% por minerales de silicato. Los más abundantes son:
- Feldespato plagioclasa (39%)
- Feldespato alcalino (12%)
- Cuarzo (12%)
- Piroxeno (11%)
- Anfíboles (5%)
- Micas (5%)
- Minerales de arcilla (5%)
Solo el 8% de la Tierra está compuesto por minerales que no son silicatos, como carbonatos, óxidos y sulfuros.
Tipos de Rocas Ígneas
Las rocas ígneas se clasifican según su contenido de sílice y otros minerales:
- Rocas félsicas: Contienen mucha sílice y, al cristalizar, dejan cuarzo libre. Ejemplos: granito, riolita.
- Rocas máficas: Contienen menos sílice y más magnesio y hierro. El cuarzo está ausente y el olivino es común. Ejemplos: gabro, basalto.
- Rocas intermedias: No tienen cuarzo ni olivino. Ejemplos: sienita, diorita, andesita.
- Rocas alcalinas: Un tipo especial de rocas intermedias o máficas con mucho álcali (especialmente sodio), que contienen minerales como la nefelina y la leucita. Ejemplos: fonolita.
- Rocas ultramáficas: Muy bajas en sílice, pero ricas en hierro y magnesio. No tienen feldespatos. Ejemplo: peridotita.
| Minerales más comunes | Félsico | Intermedio | Máfico | Ultramáfico | |
|---|---|---|---|---|---|
| Cuarzo ortoclasa (y oligoclasa), mica, hornblenda, augita |
Poco o sin cuarzo: ortoclasa hornblenda, augita, biotita |
Poco o sin cuarzo: Plagioclasa hornblenda, augita, biotita |
Sin cuarzo Plagioclasa, augita, olivino |
Sin feldespatos Augita, hornblenda, olivino |
|
| Plutónica o tipo abisal | granito | sienita | diorita | gabro | peridotita |
| Intrusiva o tipo hipoabisal | Pórfido de cuarzo | ortoclasa-porfirio | porfirita | dolerita | Picrita |
| Lavas o tipo efusivo | Riolita, obsidiana | trachyta | andesita | basalto | limburgita |
| Minerales más comunes | Alkali Feldespato, nefelina o leucita, augita, hornblenda, biotita | Soda cal Feldespato, nefelina o leucita, augita, hornblenda (olivino) | Nefelina o leucita, augita, hornblenda, olivino |
|---|---|---|---|
| Tipo plutónico | Nefelina-sienita, leucita-sienita, nefelina-porfirio | Essexita y theralita | Ijolita y missourita |
| Tipo efusivo o lavas | Fonolita, leucitophyra | Tefrita y basanita | Nefelina-basalto, leucita-basalto |
Metales Traza en el Océano
Los metales traza son elementos que se encuentran en muy pequeñas cantidades. En el océano, forman fácilmente "complejos" con otros iones. Su comportamiento químico depende de si el ambiente tiene mucho oxígeno (oxidado) o poco (reducido).
Las concentraciones de algunos metales traza, como el cadmio, cobre, molibdeno, manganeso, renio, uranio y vanadio, en los sedimentos marinos pueden indicar cómo eran las condiciones de oxígeno en los océanos en el pasado. Por ejemplo, más cadmio en los sedimentos puede significar que antes había poco oxígeno.
En el agua del océano, los metales traza disueltos se distribuyen de tres maneras:
- Tipo conservador: Tienen altas concentraciones y son poco usados por los organismos. Ejemplo: el molibdeno, que tiene un tiempo de residencia muy largo en el océano.
- Tipo nutriente: Están muy relacionados con los ciclos de la materia orgánica, especialmente con el plancton. Sus concentraciones son bajas en la superficie (donde el plancton los usa) y aumentan con la profundidad. Ejemplo: el zinc.
- Tipo barrido: Interactúan mucho con las partículas y tienen un tiempo de residencia corto. Sus concentraciones son más altas cerca del fondo, de fuentes hidrotermales o de ríos. Ejemplo: el aluminio, que llega al océano principalmente por el polvo atmosférico.
El hierro y el cobre tienen distribuciones mixtas. Son importantes nutrientes en algunas zonas del océano y se encuentran en grandes cantidades cerca de las fuentes hidrotermales.
Las técnicas electroquímicas muestran que los metales traza importantes para la vida (como el zinc, cobalto, cadmio, hierro y cobre) se unen a moléculas orgánicas en la superficie del agua de mar. Estos complejos orgánicos reducen la disponibilidad de los metales para los organismos, lo que puede ser beneficioso. Por ejemplo, el cobre puede ser tóxico para el fitoplancton, pero al formar complejos orgánicos, su toxicidad disminuye.
Galería de imágenes
-
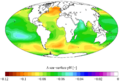
Mapa del cambio estimado del pH superficial de los océanos desde el siglo XVIII al siglo XX
-

Victor Goldschmidt (1909)
-

Abundancia de elementos del sistema solar
-

Cortes que ilustran modelos de los interiores de los planetas gigantes
Véase también
 En inglés: Geochemistry Facts for Kids
En inglés: Geochemistry Facts for Kids
- cosmoquímica
- mineralogía
- petrología
- química inorgánica
- química medioambiental
- química orgánica
- sedimentología
- suelo
- Ciclo biogeoquimico
- Ciclo geoquímico
- Tefrocronologia
