
Síndrome de Guillain-Barré para niños
Datos para niños Síndrome de Guillain-Barré o síndrome de Guillain-Barré-Strohl |
||
|---|---|---|
 Daño neuronal en el sistema nervioso periférico
|
||
| Especialidad | Neurología | |
| Síntomas | Debilidad muscular que comienza en los pies y en las manos, ausencia de reflejos, problemas de deglución | |
| Complicaciones | Dificultad para respirar, complicaciones cardíacas, parálisis de los nervios craneales. | |
| Causas | Desconocidas (posible relación con infección reciente) | |
| Diagnóstico | Basado en síntomas, electromiografía, punción lumbar | |
| Tratamiento | Inmunoglobulina por vía intravenosa, plasmaféresis, fisioterapia | |
| Frecuencia | Incidencia de 0,4 a 2 de cada 100 000 personas | |
| Tasa de letalidad | 7,5 % de los afectados | |
| Sinónimos | ||
|
||
El síndrome de Guillain-Barré (SGB) es una enfermedad neurológica poco común. Es un trastorno autoinmune, lo que significa que el sistema inmunitario del cuerpo, que normalmente nos protege de enfermedades, ataca por error a sus propios nervios. Esto afecta al sistema nervioso periférico, que son los nervios fuera del cerebro y la médula espinal.
Cuando el sistema inmunitario ataca los nervios, estos no pueden enviar señales correctamente. Esto causa debilidad muscular que suele empezar en las piernas y los brazos. A veces, puede llevar a una parálisis temporal. Aunque no se sabe la causa exacta, a menudo aparece después de una infección reciente, como una gastroenteritis o una faringitis.
Los nervios dañados no pueden transmitir bien las señales. Por eso, los músculos dejan de responder a las órdenes del cerebro. También se reciben menos señales sensoriales, lo que puede causar dificultad para sentir el calor o el dolor. La debilidad suele avanzar desde los pies hacia arriba. En algunos casos, puede afectar la respiración, y un tercio de los pacientes necesitan ayuda para respirar.
Este síndrome puede dificultar actividades como caminar o sentarse. También puede afectar el sistema nervioso autónomo, que controla funciones como la presión sanguínea y el ritmo cardíaco. La mayoría de las personas se recuperan por completo o casi por completo.
Contenido
- ¿Qué es el Síndrome de Guillain-Barré?
- Historia del Descubrimiento del SGB
- ¿Qué causa el Síndrome de Guillain-Barré?
- Síntomas del Síndrome de Guillain-Barré
- Tipos de Síndrome de Guillain-Barré
- ¿Cómo se diagnostica el SGB?
- Tratamiento del Síndrome de Guillain-Barré
- Recuperación y Pronóstico
- Galería de imágenes
- Véase también
¿Qué es el Síndrome de Guillain-Barré?
El síndrome de Guillain-Barré es una condición donde el sistema inmunitario ataca la mielina. La mielina es una capa protectora que cubre los axónes de los nervios, como el aislante de un cable eléctrico. Cuando la mielina se daña, los nervios no pueden transmitir los mensajes de forma rápida y eficiente.
¿Cómo afecta el SGB a los nervios?
Cuando los nervios no funcionan bien, los músculos no reciben las órdenes del cerebro. Esto provoca debilidad y, a veces, parálisis. También se pueden sentir hormigueos o dolor, porque las señales sensoriales no llegan bien al cerebro. Las partes del cuerpo más afectadas suelen ser las manos y los pies, ya que sus nervios son más largos.
¿Qué tan común es el SGB?
El síndrome de Guillain-Barré es raro. Afecta a entre 0.89 y 1.89 personas de cada 100,000 cada año. Es un poco más común en hombres y en personas mayores. También puede aparecer más en adolescentes y adultos jóvenes, posiblemente por ciertas infecciones.
Historia del Descubrimiento del SGB

Desde el siglo XIX, los médicos ya observaban casos de debilidad y entumecimiento que mejoraban solos. En 1859, el francés Jean Landry describió casos de debilidad que subía por el cuerpo. Él lo llamó "parálisis aguda ascendente".

En 1916, durante la Primera Guerra Mundial, tres médicos franceses, Georges Charles Guillain, Jean-Alexandre Barré y André Strohl, estudiaron a dos soldados con este tipo de parálisis. Descubrieron que estos pacientes tenían una cantidad alta de proteínas en el líquido cefalorraquídeo (el líquido que rodea el cerebro y la médula espinal), pero sin muchas células. Este hallazgo fue muy importante para entender la enfermedad.
El nombre "síndrome de Guillain-Barré" se empezó a usar en 1927. Más tarde, en 1956, el neurólogo canadiense Charles Miller Fisher describió una variante del síndrome que lleva su nombre, el síndrome de Miller Fisher. Esta variante se caracteriza por problemas en los ojos, falta de coordinación y ausencia de reflejos.
¿Qué causa el Síndrome de Guillain-Barré?
La causa exacta del SGB no se conoce por completo. Sin embargo, se sabe que es una respuesta del sistema inmunitario que ataca por error los propios nervios del cuerpo.
Infecciones y el SGB
En la mayoría de los casos (alrededor del 75%), el SGB aparece después de una infección viral o bacteriana. Se cree que el sistema inmunitario, al luchar contra la infección, se confunde y ataca partes de los nervios que se parecen a los gérmenes.
Algunas infecciones que se han relacionado con el SGB incluyen:
- Campylobacter jejuni: una bacteria que causa gastroenteritis.
- Citomegalovirus y virus de Epstein Barr: virus comunes.
- Otros virus como el del sarampión o la rubeola.
- En algunos casos, se ha asociado con el virus del Zika y el COVID-19.
Es importante saber que el SGB es un efecto secundario muy raro de estas infecciones.
¿El SGB está relacionado con las vacunas?
Se ha investigado si algunas vacunas pueden causar el SGB. En general, los estudios muestran que es extremadamente raro. Por ejemplo, se ha hablado de una posible relación con la vacuna de la gripe, pero la probabilidad de desarrollar SGB por la gripe es mayor que por la vacuna. Algunas vacunas contra el COVID-19 también han reportado casos muy raros de SGB.
Síntomas del Síndrome de Guillain-Barré
Los síntomas del SGB suelen aparecer rápidamente, en horas o días.
Debilidad y Sensaciones
- La debilidad muscular es el síntoma principal. Suele empezar en las piernas y subir hacia los brazos y la cara.
- Los pacientes pueden sentir las piernas "como de goma" o tener hormigueos y adormecimiento.
- Los reflejos, como el de la rodilla, suelen desaparecer.
- Puede haber dificultad para tragar o mover los ojos.
Otros síntomas
- En casos graves, puede haber problemas para respirar, lo que requiere ayuda médica.
- El sistema nervioso autónomo puede verse afectado, causando cambios en la presión arterial o el ritmo cardíaco.
- El dolor muscular es común, a menudo descrito como un dolor intenso después de mucho ejercicio.
Tipos de Síndrome de Guillain-Barré
Existen diferentes formas en que se presenta el SGB:
- Síndrome de Guillain-Barré "clásico" (NIAD): Es la forma más común. Causa debilidad muscular que sube por el cuerpo y pérdida de reflejos.
- Neuropatía axonal motora aguda (NAMA): Afecta principalmente a los axones de los nervios, causando debilidad muscular sin muchos problemas sensoriales.
- Síndrome de Miller-Fisher (SMF): Es una variante que se caracteriza por problemas en los movimientos de los ojos, falta de coordinación y ausencia de reflejos.
¿Cómo se diagnostica el SGB?
Diagnosticar el SGB al principio puede ser difícil porque sus síntomas se parecen a los de otras enfermedades. Los médicos realizan varias pruebas:
- Examen físico: Observan la debilidad, la simetría de los síntomas y la ausencia de reflejos.
- Punción lumbar: Se toma una muestra de líquido cefalorraquídeo de la espalda. En el SGB, este líquido suele tener muchas proteínas.
- Estudios de conducción nerviosa: Miden la velocidad con la que las señales viajan por los nervios. En el SGB, las señales son más lentas.
Tratamiento del Síndrome de Guillain-Barré
No hay una cura específica para el SGB, pero existen tratamientos que ayudan a reducir la gravedad de los síntomas y acelerar la recuperación.
Terapias principales
- Plasmaféresis: Es un procedimiento donde se extrae la sangre, se separa el plasma (la parte líquida de la sangre que contiene los anticuerpos dañinos) y luego se devuelven los glóbulos rojos y blancos al paciente. El cuerpo produce plasma nuevo rápidamente.
- Inmunoglobulina intravenosa (IVIg): Se administran altas dosis de inmunoglobulinas (proteínas que el sistema inmunitario usa para atacar gérmenes) por vía intravenosa. Estas inmunoglobulinas ayudan a bloquear los anticuerpos dañinos del paciente.
Ambos tratamientos son igual de efectivos. La inmunoglobulina suele ser más fácil de administrar.
Cuidados de apoyo
La parte más importante del tratamiento es mantener el cuerpo del paciente funcionando mientras los nervios se recuperan. Esto puede incluir:
- Ventilación mecánica: Si la respiración se ve afectada, se usa una máquina para ayudar a respirar.
- Monitoreo cardíaco: Para vigilar el ritmo del corazón.
- Fisioterapia: Es fundamental. Incluso antes de que el paciente empiece a moverse, los terapeutas mueven sus extremidades para mantener los músculos flexibles. A medida que el paciente recupera el control, la fisioterapia ayuda a fortalecer los músculos y recuperar la movilidad.
Recuperación y Pronóstico
La mayoría de las personas con SGB tienen un buen pronóstico.
- Aproximadamente el 80% de los pacientes se recuperan por completo en unos pocos meses o hasta un año.
- Entre el 5% y el 10% pueden quedar con alguna debilidad o discapacidad leve.
- La mortalidad es baja, alrededor del 4% de los pacientes.
Galería de imágenes
-
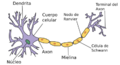
Partes de una neurona.

Véase también
 En inglés: Guillain–Barré syndrome Facts for Kids
En inglés: Guillain–Barré syndrome Facts for Kids
