
Cristalografía de rayos X para niños
La cristalografía de rayos X es una técnica científica que nos permite estudiar cómo están hechos los materiales a nivel muy pequeño, ¡como si viéramos sus átomos! Se basa en cómo los rayos X rebotan o se "difractan" cuando chocan con materiales que tienen una estructura ordenada, llamados cristales.
Los rayos X son como pequeñas ondas de luz. Cuando estas ondas chocan con los electrones que rodean a los átomos, se desvían. Los cristales tienen una estructura muy ordenada y repetitiva. Esto hace que los rayos X se desvíen de una forma especial, creando un patrón de "difracción". Este patrón es como una huella digital del material.
Con detectores especiales, podemos ver y medir este patrón. Luego, usando matemáticas, los científicos pueden crear un mapa tridimensional de los átomos y moléculas del material. Es como armar un rompecabezas para ver la forma exacta de las cosas.
Max von Laue hizo los primeros experimentos en 1912. Él, junto con William Henry Bragg y William Lawrence Bragg, sentaron las bases de esta técnica. A lo largo del siglo XX, la cristalografía de rayos X mejoró mucho. Gracias a las supercomputadoras y a máquinas especiales llamadas sincrotrones (que producen rayos X muy potentes), ahora se pueden estudiar todo tipo de moléculas. Esto incluye sales, materiales complejos, proteínas y hasta partes de las células como los ribosomas.
La forma tridimensional de las moléculas es clave para entender cómo funcionan. Por eso, la cristalografía ha ayudado mucho en campos como la química, la biología molecular, la geología, la física aplicada y la ciencia de materiales. También es útil en áreas industriales, como el desarrollo de medicamentos y el estudio de minerales.
La principal limitación de esta técnica es que solo funciona con materiales que pueden formar cristales. No se puede usar con líquidos, gases o materiales desordenados. A veces, los rayos X pueden dañar los enlaces químicos de las moléculas, especialmente en materiales biológicos. Por eso, los científicos deben tener cuidado.
Contenido
Historia de la Cristalografía de Rayos X
Los Primeros Pasos



Antes de que Wilhelm Conrad Röntgen descubriera los rayos X en 1895, ya se pensaba que los cristales eran como bloques de construcción que se repetían. Se sabía que las distancias entre estos bloques eran muy pequeñas. Esto le dio una idea al físico alemán Max von Laue: si los rayos X eran ondas, deberían crear un patrón de difracción al pasar por un cristal.
En 1912, von Laue, con la ayuda de Walter Friedrich y Paul Knipping, hizo experimentos que confirmaron esto. Poco después, los científicos británicos William Henry Bragg y su hijo William Lawrence Bragg repitieron el experimento. W.L. Bragg explicó la difracción como el rebote de los rayos X en planos paralelos dentro del cristal, lo que se conoce como la ley de Bragg.
Por sus descubrimientos, von Laue recibió el Premio Nobel de Física en 1914. Los Bragg recibieron el mismo premio un año después por sus aplicaciones prácticas. Para finales de esa década, ya se podían medir las longitudes de onda de los rayos X y se habían descubierto las estructuras de varios materiales sencillos.
Avances entre 1920 y 1960
Entre 1920 y 1960, la técnica mejoró mucho, permitiendo analizar moléculas más complejas. Se entendió mejor cómo los rayos X interactúan con los átomos. También se desarrollaron métodos matemáticos para interpretar los patrones de difracción.
En 1935, el cristalógrafo británico Arthur Patterson encontró una forma de obtener las distancias entre átomos directamente de los datos experimentales, lo que fue un gran avance. En las décadas de 1940 a 1960, surgieron nuevos métodos para encontrar estructuras, incluso para moléculas orgánicas. Un gran logro fue la determinación de la estructura de la vitamina B12 en 1957, liderado por Dorothy Hodgkin.
La Era de las Macromoléculas: 1960-1980
Las décadas de 1960 y 1970 fueron importantes para la cristalografía de proteínas y otras moléculas biológicas. En 1962, se otorgaron dos Premios Nobel por descubrimientos hechos con esta técnica:
- El premio Nobel de Química a Max Perutz y John Kendrew por sus estudios de las proteínas hemoglobina y mioglobina.
- El Premio Nobel de Medicina a Francis Crick, James Watson y Maurice Wilkins por descubrir la estructura de doble hélice del ADN.
En 1964, Dorothy Hodgkin también ganó el Premio Nobel de Química por determinar la estructura de la penicilina y la vitamina B12.
El desarrollo de las computadoras digitales fue clave. Antes, los cálculos se hacían a mano, lo que era muy lento. Con las computadoras, se pudo medir y analizar los datos mucho más rápido. Sin embargo, en ese tiempo, aún faltaban fuentes de rayos X más potentes y mejores detectores.
Expansión y Madurez: 1980-2010

Desde los inicios, los tubo de rayos X fueron la principal fuente de rayos X. Pero en los años 70, se empezó a usar la radiación de sincrotrón, que es mucho más intensa. Los sincrotrones son grandes instalaciones que producen rayos X muy potentes.
En el siglo XXI, aparecieron los primeros láseres de electrones libres de rayos X. Estos producen pulsos de luz extremadamente cortos y brillantes, lo que permite observar cambios en los cristales en tiempos muy, muy rápidos (femtosegundos). También se crearon nuevos detectores que generan imágenes digitales de los patrones de difracción al instante, reemplazando las antiguas placas fotográficas.
Todos estos avances, junto con el aumento de la capacidad de las computadoras, han permitido estudiar un número enorme de estructuras. Por ejemplo, a principios de 2013, había más de 88,000 estructuras de macromoléculas orgánicas en el Banco de Datos de Proteínas. Un gran logro fue la obtención de la estructura del ribosoma en el año 2000, una parte esencial de la célula que fabrica proteínas.
Cómo Funciona la Cristalografía
Cristales y sus Redes

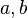 y
y  .
.
Los cristales están hechos de moléculas que se repiten una y otra vez en el espacio. Podemos imaginar un cristal como una red tridimensional donde cada punto es igual. La parte más pequeña que se repite para formar el cristal se llama celda unidad. Es como el ladrillo básico de una pared.
La celda unidad se define por la longitud de sus lados (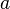 ,
, 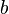 , ) y los ángulos entre ellos (
, ) y los ángulos entre ellos (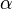 ,
,  ,
, 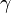 ). El objetivo de la cristalografía es encontrar la posición exacta de cada átomo dentro de esta celda unidad.
). El objetivo de la cristalografía es encontrar la posición exacta de cada átomo dentro de esta celda unidad.
Los Rayos X

Los rayos X son un tipo de radiación electromagnética, como la luz visible, pero con una longitud de onda mucho más corta. Para la cristalografía, se usan rayos X con una longitud de onda de aproximadamente 0.1 nanómetros, que es similar al tamaño de un átomo.
Cuando los rayos X atraviesan un material, interactúan con los electrones de los átomos. Esta interacción hace que la onda de rayos X cambie su "fase", es decir, su posición en el ciclo de la onda. Este cambio de fase contiene información sobre dónde están los átomos.
Cómo se Dispersan los Rayos X
La difracción en un cristal ocurre porque los rayos X rebotan en los átomos del cristal. Es como si cada átomo fuera un pequeño espejo. Los rayos X dispersados salen en un ángulo específico.
La forma en que los rayos X se dispersan por un grupo de átomos se describe con algo llamado "factor de estructura". Este factor nos dice cómo se suman los rayos que rebotan en cada átomo. Depende del número de electrones del átomo y del ángulo en que inciden los rayos X.
La Difracción en Cristales


La Ley de Bragg

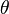 . Si la diferencia en el camino recorrido por las ondas es un múltiplo entero de la longitud de onda, las ondas se amplifican y se observa difracción.
. Si la diferencia en el camino recorrido por las ondas es un múltiplo entero de la longitud de onda, las ondas se amplifican y se observa difracción.La difracción ocurre cuando las ondas de rayos X que rebotan en diferentes partes del cristal se suman de forma constructiva. Imagina que tienes muchas olas en el mar. Si dos olas se encuentran y sus crestas coinciden, se hacen más grandes. Si una cresta se encuentra con un valle, se anulan.
En un cristal, los rayos X que rebotan en diferentes planos de átomos deben estar "en fase" (sus crestas deben coincidir) para que se vea un punto brillante en el detector. La condición para que esto suceda se describe con la ley de Bragg:
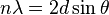
Aquí, 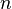 es un número entero,
es un número entero, 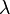 es la longitud de onda de los rayos X,
es la longitud de onda de los rayos X, 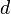 es la distancia entre los planos de átomos en el cristal, y es el ángulo de incidencia de los rayos X.
es la distancia entre los planos de átomos en el cristal, y es el ángulo de incidencia de los rayos X.
Índices de Miller

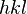 y la orientación de los planos en el sistema de coordinadas definido por aquella.
y la orientación de los planos en el sistema de coordinadas definido por aquella.Para identificar los diferentes planos de átomos en un cristal, se usan tres números llamados índices de Miller: 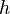 ,
, 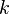 y
y 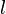 . Estos números nos dicen cómo se orientan los planos dentro de la celda unidad. Cada punto en el patrón de difracción corresponde a un conjunto específico de planos de Miller.
. Estos números nos dicen cómo se orientan los planos dentro de la celda unidad. Cada punto en el patrón de difracción corresponde a un conjunto específico de planos de Miller.
El Problema de las Fases
El objetivo final es obtener un mapa de la densidad de electrones en el cristal, que nos dice dónde están los átomos. Para esto, necesitamos conocer no solo la intensidad de los puntos de difracción (que se mide fácilmente), sino también la "fase" de cada onda difractada. La fase es como la posición inicial de la onda. El problema es que no podemos medir la fase directamente. Esto se conoce como el "problema de las fases".
Existen varios métodos para resolver este problema:
- Métodos de Patterson: Usan la intensidad de los puntos de difracción para encontrar las distancias entre pares de átomos.
- Métodos directos: Usan propiedades matemáticas de la densidad de electrones para adivinar las fases. Funcionan bien para moléculas pequeñas.
- Dispersión anómala: Aprovecha que algunos átomos pesados interactúan de forma especial con los rayos X, cambiando la fase de las ondas.
- Reemplazamiento isomorfo: Consiste en añadir átomos pesados a la molécula que se quiere estudiar. Las diferencias en la difracción ayudan a encontrar las fases.
- Reemplazamiento molecular: Se usa cuando ya se conoce la estructura de una molécula similar. Se usa esa estructura conocida como punto de partida para adivinar las fases de la nueva molécula.
Refinamiento de la Estructura
Una vez que se tienen las fases, se puede crear un primer modelo de la estructura de la molécula. Este modelo puede no ser perfecto. El "refinamiento" es un proceso para mejorar este modelo. Se ajustan las posiciones de los átomos y otros parámetros hasta que el patrón de difracción calculado a partir del modelo sea lo más parecido posible al patrón medido experimentalmente.
Se usa un valor llamado "factor R" para saber qué tan bueno es el modelo. Cuanto más bajo sea el factor R, mejor es el modelo.
Cristalización

Para poder estudiar un material con cristalografía de rayos X, primero necesitamos que forme un cristal de buena calidad. A veces, los cristales se encuentran en la naturaleza (como los minerales), pero a menudo hay que crearlos en el laboratorio.
Un método común es la "difusión de vapor". Se prepara una solución con el material y se deja que el disolvente se evapore lentamente, haciendo que el material se concentre y forme cristales. Otro método es la cristalización en geles, útil para materiales que no se disuelven bien.
El tamaño del cristal es importante. Los cristales muy grandes pueden absorber demasiados rayos X, y los muy pequeños difractan débilmente. Para materiales orgánicos, se prefieren cristales de unas décimas de milímetro. Con las fuentes de rayos X modernas, se pueden usar cristales aún más pequeños, de unas veinte micras o menos.
Cómo se Obtienen los Datos
Fuentes de Rayos X

Para los experimentos, necesitamos una fuente de rayos X. Hay dos tipos principales:
- Tubos de rayos X: Son más pequeños y económicos. Funcionan acelerando electrones que chocan contra un metal, produciendo rayos X.
- Sincrotrones: Son instalaciones mucho más grandes y potentes. Aceleran electrones a velocidades cercanas a la luz y los desvían con campos magnéticos, lo que produce rayos X muy intensos. Estos son necesarios para cristales muy pequeños o con átomos ligeros.
En los últimos años, también han aparecido los láseres de electrones libres de rayos X. Estos producen pulsos de rayos X extremadamente cortos y brillantes, lo que permite estudiar procesos muy rápidos y cristales diminutos.
Detectores
Para registrar los patrones de difracción, se usan detectores. Antes se usaba película fotográfica, pero ahora hay detectores electrónicos mucho más sensibles y rápidos. Estos detectores "de área" pueden generar una imagen digital del patrón de difracción al instante.
Métodos de Difracción
Hay varias formas de medir la difracción de rayos X:
Método de Laue

En este método, se usa un haz de rayos X con muchas longitudes de onda diferentes. Esto permite obtener muchas reflexiones sin tener que mover el cristal. Es muy útil para estudiar reacciones químicas rápidas o cambios en materiales bajo diferentes condiciones.
Métodos de Rotación de Cristal


Aquí, se usa un haz de rayos X de una sola longitud de onda y se hace girar el cristal. A medida que el cristal rota, diferentes planos de átomos se alinean para difractar los rayos X, y el detector registra los patrones. Gracias a las computadoras, este es el método más usado hoy en día.
Método de Polvo Cristalino

En este método, el material se pulveriza hasta formar muchos microcristales pequeños. Como están en todas las orientaciones posibles, se pueden medir todas las reflexiones en un solo patrón, que se ve como círculos. Es útil cuando no se pueden obtener cristales grandes, por ejemplo, para identificar minerales en una muestra.
Cristalografía en Serie
Esta es una técnica más reciente. Consiste en hacer pasar una corriente de microcristales (muy pequeños, de micras) a través de un haz de rayos X. Un detector registra continuamente los datos. La idea es obtener la difracción de los cristales antes de que los rayos X los dañen. Esto es especialmente útil para estudiar proteínas de membrana y virus.
Aplicaciones de la Cristalografía
La cristalografía tiene muchísimas aplicaciones en diversas ciencias: mineralogía, química, biología molecular, farmacología, geología, física aplicada y ciencia de materiales. También es útil en campos como la ciencia forense y la historia del arte, para identificar materiales en obras antiguas.
Premios Nobel

Muchos descubrimientos importantes hechos con cristalografía de rayos X han sido reconocidos con el Premio Nobel. Algunos ejemplos incluyen:
- En 1962, Kendrew y Perutz (Química) y Crick, Watson y Wilkins (Medicina) por sus trabajos con proteínas y el ADN.
- En 1964, Dorothy Hodgkin (Química) por la estructura de la penicilina y la vitamina B12.
- En 2009, Ramakrishnan, Steitz y Yonath (Química) por estudiar la estructura y función del ribosoma.
- También se han dado Premios Nobel por el desarrollo de la propia técnica, como a Max von Laue y los hermanos Bragg.
Análisis de Minerales
La cristalografía de rayos X se usa para saber qué minerales hay en muestras de suelo o rocas. Cada mineral tiene un patrón de difracción único. El método de polvo es muy útil para esto, ya que se pueden comparar los patrones con una base de datos conocida.
El vehículo explorador Curiosity en Marte tiene un difractómetro de rayos X. En 2012, envió las primeras imágenes de difracción desde Marte, mostrando la presencia de minerales como feldespato y olivino en el suelo marciano.
Diseño de Medicamentos
Desde finales del siglo XX, la cristalografía ha ayudado a diseñar nuevos medicamentos. Al conocer la estructura tridimensional de las proteínas, los científicos pueden entender cómo interactúan con otras moléculas. Esto permite diseñar medicamentos que se ajusten perfectamente a una proteína específica para bloquear o activar su función. Un ejemplo es el vemurafenib, un medicamento diseñado para tratar un tipo de cáncer de piel.
Comparación con Otras Técnicas
Además de los rayos X, también se pueden estudiar cristales usando la difracción de neutrones o de electrones. Aunque son similares, tienen algunas diferencias:
- Difracción de electrones: Los electrones interactúan fuertemente con los átomos, lo que permite estudiar muestras muy pequeñas. También se pueden obtener las fases directamente de las imágenes de un microscopio electrónico.
- Difracción de neutrones: Los neutrones interactúan solo con el núcleo de los átomos. Son muy buenos para ver átomos de hidrógeno, que son difíciles de ver con rayos X. Además, los neutrones no dañan las moléculas durante el experimento.
Limitaciones
La principal limitación de la cristalografía de rayos X es que solo funciona con materiales que pueden formar cristales. No se puede usar para estudiar gases, líquidos o sólidos desordenados. Las moléculas muy grandes también pueden ser difíciles de cristalizar.
Otra limitación es que el modelo que se obtiene es un promedio de las posiciones de los átomos en el cristal. Si una molécula se mueve mucho o tiene partes desordenadas, esas partes pueden no verse bien en el mapa de densidad electrónica. Esto es un problema para estudiar enzimas, por ejemplo, si su parte activa es muy móvil.
Además, los rayos X pueden dañar o destruir las moléculas del cristal durante el experimento. Esto es un problema grave en cristales de proteínas, que son muy sensibles. Para reducir este daño, los experimentos con moléculas biológicas suelen hacerse a temperaturas muy bajas. Los láseres de electrones libres de rayos X, con sus pulsos ultracortos, permiten obtener datos antes de que el daño sea significativo.
Véase también
 En inglés: X-ray crystallography Facts for Kids
En inglés: X-ray crystallography Facts for Kids
- Cristalografía
- Difracción de electrones
- Difracción de neutrones
- Espectroscopia de rayos X
- Resonancia magnética nuclear
