
Progeria para niños
Datos para niños Progeria |
||
|---|---|---|
 Niño con síndrome de progeria. Arriba a la derecha: Núcleo celular sano; Abajo a la derecha: Núcleo celular progérico.
|
||
| Especialidad | endocrinología | |
| Síntomas | Retraso en el crecimiento, baja estatura, cara pequeña, caída del cabello. | |
| Complicaciones | Enfermedad cardíaca, accidente cerebrovascular, luxación de cadera, venas inusualmente prominentes en cuero cabelludo,pérdida de la capa de grasa subcutánea (tejido adiposo subcutáneo), defectos ungueales, rigidez articular, defectos esqueléticos y otras anomalías. | |
| Diagnóstico diferencial | Síndrome de Hallermann-Streiff, síndrome de Gottron, síndrome de Wiedemann-Rautenstrauch. | |
| Sinónimos | ||
| Síndrome de Werner. Envejecimiento prematuro o algunos casos de extrema rareza de envejecimiento prematuro en un periodo de 6 años de vida. Envejecimiento precoz. Progeria del adulto. Síndrome de Joseph. | ||
La progeria (del griego pro, que significa 'hacia' o 'a favor de', y geron, que significa 'viejo') es una enfermedad genética muy poco común. Esta condición hace que el cuerpo de los niños envejezca mucho más rápido de lo normal. Generalmente, los primeros signos aparecen entre el primer y segundo año de vida.
Se calcula que afecta a uno de cada 20 millones de recién nacidos. No hay pruebas de que sea más común en un sexo que en otro. Sin embargo, se ha observado con mayor frecuencia en personas de etnia blanca (97%). La progeria afecta a varios órganos y tejidos del cuerpo. Esto incluye los huesos, los músculos, la piel, la grasa debajo de la piel y los vasos sanguíneos.
Los niños con progeria suelen tener baja estatura y un cráneo grande. También pueden perder el cabello, tener la piel seca y arrugada, poca grasa bajo la piel y articulaciones rígidas. Actualmente, no hay una cura conocida ni un tratamiento que detenga la enfermedad. La esperanza de vida promedio para quienes la padecen es de unos trece años. Sin embargo, algunos pacientes han vivido un poco más de veinte años.
La forma más grave de progeria se llama «síndrome de Hutchinson-Gilford». Recibe su nombre de Jonathan Hutchinson, quien la describió por primera vez en 1886. También se nombra en honor a Hastings Gilford, quien realizó más estudios sobre su desarrollo en 1904.
Contenido
¿Qué es la Progeria?
La progeria es una condición genética que provoca un envejecimiento acelerado. Esto significa que los niños que la tienen muestran características de personas mayores a una edad muy temprana. Es una enfermedad muy rara que afecta a muy pocos niños en el mundo.
¿Cómo se manifiesta la Progeria en los niños?
Los niños con progeria presentan varias características físicas que se asemejan al envejecimiento. Estas características suelen aparecer durante los primeros años de vida.
- Estatura baja.
- Piel seca y arrugada.
- Calvicie prematura (pérdida de cabello).
- Canas en el cabello desde la infancia.
- Ojos que parecen más grandes de lo normal.
- Cráneo de gran tamaño.
- Venas en la cabeza que se ven muy marcadas.
- Ausencia de cejas y pestañas.
- Nariz grande y con forma de gancho.
- Mentón retraído (hacia atrás).
- Problemas del corazón.
- Pecho angosto, con costillas que se notan mucho.
- Extremidades (brazos y piernas) delgadas y con poca masa.
- Estrechamiento de las arterias que van al corazón.
- Articulaciones grandes y rígidas.
- Manchas en la piel similares a las de la vejez.
- Problemas en los dientes.
- Debilitamiento de algunos huesos, como las clavículas y los dedos.
- Osteoporosis (huesos más frágiles).
- Endurecimiento de las arterias (arterioesclerosis).
- Problemas de salud que normalmente aparecen en la vejez, como la artritis.
¿Qué causa la Progeria?
La progeria es causada por un cambio (mutación) en un gen llamado LMNA. Este gen es muy importante porque produce proteínas llamadas láminas A y C. Estas proteínas forman parte de la "lámina nuclear", una estructura que ayuda a mantener la forma y el funcionamiento del núcleo de nuestras células.
Cuando el gen LMNA tiene esta mutación, produce una proteína alterada llamada progerina. La progerina no funciona correctamente y afecta la forma en que el núcleo de la célula se organiza. Esto daña la integridad de los tejidos conectivos del cuerpo, que son como el "pegamento" que une y soporta nuestros órganos.
¿Cómo afecta la progerina a las células?
Las células con progerina tienen núcleos con formas extrañas, como si tuvieran "bolsas" o "lóbulos". También tienen problemas en la organización de su material genético (cromatina). Además, estas células tienen dificultades para reparar su ADN cuando se daña.
La energía de las células y la progeria
La progerina también provoca que se acumulen en las células unas estructuras llamadas mitocondrias que no funcionan bien. Las mitocondrias son como las "centrales de energía" de la célula. Cuando no funcionan correctamente, producen menos energía y más sustancias dañinas. Esto contribuye a que las células envejezcan más rápido. Los científicos siguen investigando por qué ocurre esto y cómo se puede evitar.
¿Cómo se diagnostica la Progeria?
Al nacer, los bebés con progeria no suelen mostrar características especiales. A veces, se pueden notar una forma particular de la nariz o cambios en la piel. Los síntomas más claros aparecen durante el primer año de vida. Pueden incluir un retraso en el crecimiento, pérdida de cabello y cambios en la piel. Hacia el segundo año, la caída del cabello se hace más evidente.
El diagnóstico de la progeria se basa principalmente en la observación de los síntomas. Los médicos buscan una combinación de "criterios mayores" y "signos" que suelen estar presentes.
Criterios principales para el diagnóstico
Los criterios mayores incluyen:
- Cara delgada.
- Pérdida de cabello.
- Venas muy visibles en la cabeza.
- Ojos grandes.
- Mandíbula pequeña.
- Dientes que salen de forma anormal o tardía.
- Pecho con forma de campana o pera.
- Clavículas cortas.
- Piernas arqueadas.
- Brazos delgados con articulaciones prominentes.
- Baja estatura y bajo peso para la edad.
- Desarrollo incompleto en la pubertad.
- Poca grasa debajo de la piel.
Los signos que suelen estar presentes son:
- Piel endurecida.
- Pérdida generalizada de cabello.
- Orejas prominentes sin lóbulos.
- Nariz con forma de gancho.
- Labios delgados.
- Paladar alto.
- Voz aguda.
- Uñas con problemas.
En resumen, el diagnóstico se confirma en niños que muestran los primeros signos y cumplen con todos los criterios mayores. Actualmente, no existe una prueba de laboratorio definitiva que certifique el diagnóstico de progeria.
¿Con qué otras condiciones se puede confundir la Progeria?
Es importante diferenciar la progeria de otros síndromes similares. Algunos de ellos son:
- El síndrome de Werner, a veces llamado "progeria de los adultos". Es más común que la progeria infantil.
- El síndrome de Mulvill-Smith, que causa retraso en el crecimiento antes del nacimiento, baja estatura y cabeza pequeña.
- El síndrome de Cockayne, cuyos síntomas aparecen más tarde en la vida. Incluye sensibilidad de la piel al sol y problemas en los ojos.
¿Cuál es el pronóstico de la Progeria?
La esperanza de vida promedio para los niños con progeria es de unos 13 años. Aunque algunos pueden vivir entre 7 y 27 años, es raro que sobrevivan más allá de la adolescencia. La mayoría de los casos de fallecimiento (más del 80%) se deben a problemas del corazón y los vasos sanguíneos. Esto incluye el endurecimiento de las arterias y problemas de circulación.
Actualmente, no hay una cura ni un medicamento que detenga la progeria. Sin embargo, los tratamientos se enfocan en prevenir o retrasar las complicaciones, especialmente las del corazón. Se usan medicamentos como la aspirina en dosis bajas y dietas especiales. También se han probado tratamientos con hormona de crecimiento.
Después de descubrir el gen que causa la enfermedad, se ha investigado un tipo de medicamento contra el cáncer. Estos medicamentos, llamados inhibidores de la farnesiltransferasa (FTIs), han mostrado resultados prometedores en estudios con ratones. Desde 2007, se han realizado pruebas clínicas con pacientes usando un FTI llamado Lonafarnib.
Recientemente, se ha descubierto que una proteína llamada CRM1 está muy activa en las células de pacientes con progeria. Esto causa problemas en el transporte de proteínas dentro y fuera del núcleo de la célula. Se ha visto que al bloquear esta proteína, se pueden mejorar los síntomas de envejecimiento celular y la función de las mitocondrias. Estos resultados están siendo validados para su uso en humanos.
Aunque se ha avanzado mucho en la comprensión de la progeria, aún no hay una cura. El hecho de que los pacientes vivan pocos años dificulta la realización de estudios a largo plazo.
Casos conocidos de Progeria
A lo largo de los años, algunos casos de progeria han sido conocidos públicamente, ayudando a crear conciencia sobre esta condición.
- En 1981, el programa de televisión That's Incredible! mostró el caso de Fransie Geringer, un niño sudafricano de 8 años con progeria. Él visitó a Mickey Hays, otro niño con la misma condición, y juntos fueron a Disneylandia.
- También está el caso de Hayley Okines, una niña que apareció en un documental de National Geographic Channel cuando tenía 13 años.
- El documental Life According to Sam (La vida según Sam, 2013), de Sean Fine y Andrea Nix, se centró en la historia de Sam Berns. Este documental explicó qué es la progeria, cómo se descubrieron sus causas y los esfuerzos para conseguir la aprobación del primer medicamento para tratar sus síntomas, llamado Lonafarnib.
- En octubre de 2024, falleció Sammy Basso a los 28 años. Sammy fue un joven con una notable capacidad académica que ayudó a crear conciencia sobre la progeria a través de múltiples apariciones, incluyendo un documental de National Geographic. En 2018, obtuvo un doctorado en Ciencias Naturales en la Universidad de Padua, donde investigó posibles terapias para retrasar el desarrollo de la progeria. En 2019, fue reconocido como Caballero de la Orden del Mérito por la República de Italia por sus contribuciones científicas.
Galería de imágenes
-
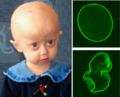
Niño con síndrome de progeria. Arriba a la derecha: Núcleo celular sano; Abajo a la derecha: Núcleo celular progérico.
Véase también
 En inglés: Progeria Facts for Kids
En inglés: Progeria Facts for Kids
