
Síndrome de Werner para niños
Datos para niños Síndrome de Werner |
||
|---|---|---|
|
El síndrome de Werner tiene un patrón autosómico recesivo
|
||
| Especialidad | endocrinología | |

El síndrome de Werner es una condición genética muy poco común. Se caracteriza por un envejecimiento acelerado del cuerpo. Fue nombrado así por el científico alemán Otto Werner, quien lo descubrió al observar a cuatro hermanos que mostraban signos de envejecimiento antes de tiempo.
Esta condición es muy rara, afectando a menos de 1 de cada 100,000 personas en el mundo. Se han reportado alrededor de 1,300 casos. Las personas con síndrome de Werner pueden experimentar pérdida de cabello, cataratas en ambos ojos, un tipo de diabetes, osteoporosis (huesos débiles) y problemas en las arterias. También pueden aparecer úlceras en la piel, especialmente en los tobillos, y una pérdida de grasa debajo de la piel. Quienes tienen este síndrome tienen un riesgo más alto de desarrollar ciertos tipos de cáncer, como los sarcomas. La edad promedio de fallecimiento para las personas afectadas es entre los 46 y 48 años.
Contenido
¿Cómo se hereda el síndrome de Werner?
Este síndrome se hereda de una manera específica llamada autosómico recesivo. Esto significa que una persona debe heredar dos copias del gen alterado (una de cada padre) para desarrollar la condición. Si solo hereda una copia, será un "portador" y no mostrará los síntomas.
El gen WRN y sus cambios
Los síntomas de esta condición no suelen aparecer hasta que la persona tiene alrededor de treinta años. El gen responsable, llamado WRN, se encuentra en el cromosoma 8. En las personas sanas, el gen WRN produce una proteína importante. Sin embargo, en las personas con síndrome de Werner, este gen tiene cambios (llamados mutaciones). Estos cambios impiden que la proteína funcione correctamente.
La proteína que produce el gen WRN se llama RecQl2. Esta proteína es como una herramienta que ayuda a nuestras células a manejar el ADN. Participa en procesos clave como la reparación del ADN, la copia del ADN (replicación) y la reorganización del ADN.
¿Qué le pasa al cuerpo?
Las células de las personas con síndrome de Werner muestran varios problemas. Por ejemplo, su genoma (todo su ADN) es menos estable. Esto significa que el ADN puede dañarse más fácilmente. También tienen dificultades para reparar el ADN cuando se rompe.
El papel de los telómeros
Nuestras células tienen unas "tapas" protectoras al final de sus cromosomas, llamadas telómeros. Cada vez que una célula se divide, los telómeros se acortan un poco. En las personas con síndrome de Werner, los telómeros se acortan mucho más rápido de lo normal. Esto hace que las células envejezcan y dejen de dividirse antes de tiempo, lo que contribuye a los signos de envejecimiento acelerado.
¿Qué tan común es?
La frecuencia del síndrome de Werner varía en diferentes partes del mundo. En Japón, por ejemplo, es más común, afectando a 1 de cada 20,000 a 40,000 personas. En América del Norte, se estima que afecta a 1 de cada 200,000 personas.
¿Cómo se diagnostica?
El diagnóstico del síndrome de Werner se basa en la observación de los síntomas característicos. Sin embargo, para confirmar el diagnóstico, se puede realizar una prueba genética. Esta prueba analiza el gen WRN para encontrar las mutaciones. Esto se puede hacer en etapas tempranas, incluso antes de que aparezcan todos los síntomas, o si se sabe que hay familiares afectados.
¿Existe un tratamiento?
Actualmente, no existe una cura para el síndrome de Werner. El tratamiento se enfoca en aliviar los síntomas y mejorar la calidad de vida de las personas afectadas. Esto incluye manejar la diabetes, tratar las úlceras de la piel y controlar otros problemas de salud que puedan surgir. Los científicos están investigando posibles tratamientos futuros.
Consejos para las familias
Si no hay antecedentes de síndrome de Werner en la familia, es muy poco probable que sea una preocupación. Sin embargo, si hay un familiar afectado o si la familia tiene orígenes en el norte de Japón, se podría recomendar un análisis genético.
Si ambos padres son portadores del gen alterado (es decir, tienen una copia del gen pero no la condición), cada hijo tiene un 25% de probabilidad de heredar el síndrome, un 50% de ser portador (como los padres) y un 25% de no tener el gen alterado en absoluto.
El síndrome de Werner generalmente comienza a mostrarse a partir de los 30 años. Como no tiene cura, el objetivo principal es brindar apoyo y cuidado para manejar los síntomas.
Galería de imágenes
-
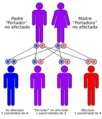
El síndrome de Werner tiene un patrón autosómico recesivo
Véase también
 En inglés: Werner syndrome Facts for Kids
En inglés: Werner syndrome Facts for Kids
