
Tumor de Wilms para niños
Datos para niños Tumor de Wilms |
||
|---|---|---|
 Células tumorales (las células estrelladas) de un nefroblastoma. Microscopía electrónica.
|
||
| Especialidad | oncología | |
| Sinónimos | ||
|
||
El tumor de Wilms o nefroblastoma es un tipo de tumor que se forma en el riñón. Es el segundo tumor más común en el abdomen de los niños, después de otro tipo de tumor que afecta las glándulas suprarrenales.
Este tumor suele aparecer en la infancia, afectando a 1 de cada 200,000 a 250,000 niños. Es muy raro en niños mayores de 8 años y en recién nacidos. Recibe su nombre de Max Wilms (1867-1918), un cirujano alemán que lo describió por primera vez.
Alrededor del 75% de los casos ocurren en niños que no tienen otros problemas de salud. El 25% restante se asocia con algunas condiciones de desarrollo, como problemas en las vías urinarias, la ausencia de la parte de color del ojo (llamada iris) o un crecimiento desproporcionado de un lado del cuerpo (hemihipertrofia).
Este tumor responde muy bien al tratamiento. Las posibilidades de que un niño se recupere y viva al menos 5 años después del diagnóstico son muy altas, superando el 90%.
Contenido
¿Cómo se desarrolla el tumor de Wilms?
La mayoría de los tumores de Wilms afectan solo un riñón. En menos del 5% de los casos, afectan a ambos riñones.
Estos tumores suelen crecer en la parte superior del riñón. Generalmente, están cubiertos por una capa protectora y tienen muchos vasos sanguíneos. No suelen crecer más allá de la mitad del abdomen. Si el tumor se extiende a otras partes del cuerpo, lo más común es que lo haga a los pulmones.
Un tumor de Wilms puede romperse. Si esto ocurre, existe un riesgo importante de sangrado y de que las células del tumor se extiendan por el abdomen. Si el tumor se rompe, es muy importante que un cirujano con experiencia lo extirpe rápidamente.
¿De qué está hecho el tumor?
Un tumor de Wilms está formado por tres tipos principales de células:
- Blastema: Son células que se parecen a las que se encuentran en las primeras etapas de desarrollo de un bebé.
- Mesénquima: Es un tipo de tejido conectivo.
- Epitelio: Son las células que recubren las superficies del tumor.
El tumor de Wilms es un tumor que contiene estas células inmaduras, junto con tejido que se parece a los glomérulos y túbulos del riñón, rodeados por un tejido de soporte. Este tejido de soporte puede incluir músculo, cartílago, hueso, grasa y tejido fibroso. El tumor suele presionar el tejido normal del riñón.
Los tumores de Wilms se clasifican en dos grupos según sus características:
- Favorable: Contiene células bien desarrolladas.
- Anaplásico: Contiene células con un desarrollo escaso.
El riesgo de desarrollar un tumor de Wilms es mayor en niños con ciertas condiciones de salud, como la aniridia, la criptorquidia, la hipospadias y otros problemas congénitos de las vías urinarias. También es más común en niños con síndromes específicos como el síndrome de Beckwith Wiedemann, el síndrome de Denys-Drash, el síndrome de Bloom y el síndrome WAGR.
¿Qué causa el tumor de Wilms?

Se ha encontrado una relación genética en muchos casos de tumor de Wilms. Se cree que la causa puede estar relacionada con un gen llamado WT1, que se encuentra en el cromosoma 11. Cuando este gen está alterado, aumenta el riesgo de que el niño desarrolle este tumor y otras condiciones de salud.
Otros genes y cromosomas también pueden estar involucrados en el desarrollo de este tumor. Por ejemplo, se ha visto que cambios en el gen CTNNB1 son comunes en pacientes con tumor de Wilms que también tienen cambios en el gen WT1. Además, se ha descubierto que un gen en el cromosoma X, llamado WTX, está inactivo en algunos pacientes. Sin embargo, en la mayoría de los casos, no se encuentran cambios en ninguno de estos genes.
¿Cuáles son los síntomas del tumor de Wilms?

En el 95% de los casos, el síntoma principal es una masa o bulto que se puede sentir en el abdomen. La mayoría de los niños con un tumor de Wilms no se sienten muy enfermos en general.
Algunos de los síntomas que pueden aparecer son:
- Dolor abdominal
- Un bulto en el abdomen
- Sangre en la orina (en menos del 10% de los casos)
- Fiebre
- Pérdida del apetito
- Náuseas o vómitos
- Cansancio (astenia)
- Presión arterial elevada
- Estreñimiento
¿Cómo se diagnostica el tumor de Wilms?
Si un médico encuentra un bulto en el abdomen de un niño durante un examen, se recomienda hacer un ultrasonido en las siguientes 24 horas. Este estudio debe ser realizado por un especialista en imágenes con experiencia en niños. Si el ultrasonido no aclara el origen del bulto, se puede hacer una tomografía.
Si se confirma que el tumor proviene del riñón, el niño debe ser referido de inmediato a un oncólogo pediatra (un médico especialista en tumores en niños) que tenga experiencia en cirugía.
¿Cómo se trata el tumor de Wilms?
Antes de iniciar el tratamiento, es muy importante saber qué tan extendido está el tumor. La cirugía para extirpar el tumor es necesaria, y a veces también se extirpan tejidos cercanos que puedan estar afectados.
El uso de radioterapia (tratamiento con radiación) y quimioterapia (tratamiento con medicamentos) ha mejorado mucho las posibilidades de recuperación de los pacientes. Gracias a estos tratamientos, las tasas de supervivencia están entre el 80% y el 90%.
Etapas del tumor de Wilms
La etapa del tumor se determina combinando los resultados de los estudios de imagen, lo que se encuentra al examinar el tumor en el laboratorio y si el tumor se puede extirpar con cirugía. El tratamiento se adapta a cada etapa:
Etapa I (43% de los pacientes)
En esta etapa, el tumor está limitado al riñón y se puede extirpar por completo. La superficie del riñón está intacta, el tumor no se ha roto ni se ha tomado una muestra antes de la cirugía. No hay células tumorales en los vasos sanguíneos del riñón ni quedan restos de tumor después de la cirugía.
- Tratamiento: Cirugía para extirpar el riñón afectado (nefrectomía) y 18 semanas de quimioterapia.
- Pronóstico: El 98% de los niños sobreviven al menos 4 años. Si el tumor es de tipo anaplásico, la supervivencia a los 4 años es del 85%.
Etapa II (23% de los pacientes)
En esta etapa, el tumor se ha extendido un poco más allá del riñón, pero se puede extirpar por completo. No quedan restos de tumor después de la cirugía. Puede que el tumor se haya extendido fuera del riñón o a los vasos sanguíneos del riñón. También puede que se haya tomado una muestra del tumor antes de la cirugía o que el tumor se haya extendido un poco durante la cirugía, pero solo en la zona cercana.
- Tratamiento: Cirugía para extirpar el riñón afectado, radioterapia en el abdomen y 24 semanas de quimioterapia.
- Pronóstico: La supervivencia a los 4 años es del 95%. Si el tumor es de tipo anaplásico, la supervivencia a los 4 años es del 70%.
Etapa III (23% de los pacientes)
En esta etapa, el tumor no se puede extirpar completamente con cirugía. Puede haber células tumorales en los ganglios linfáticos cercanos, o quedar células tumorales en los bordes de la cirugía. También puede haber extensión del tumor en las superficies del abdomen antes o durante la cirugía.
- Tratamiento: Radioterapia en el abdomen, 24 semanas de quimioterapia y cirugía para extirpar el riñón después de que el tumor se haya reducido.
- Pronóstico: La supervivencia a los 4 años es del 95%. Si el tumor es de tipo anaplásico, la supervivencia a los 4 años es del 70%.
Etapa IV (10% de los pacientes)
En esta etapa, el tumor se ha extendido a otras partes del cuerpo a través de la sangre, como los pulmones, el hígado, los huesos o el cerebro. También puede haber células tumorales en los ganglios linfáticos del abdomen y la pelvis que están lejos del riñón.
- Tratamiento: Cirugía para extirpar el riñón afectado, radioterapia en el abdomen, 24 semanas de quimioterapia y radioterapia en las zonas donde se ha extendido el tumor, si es necesario.
- Pronóstico: La supervivencia a los 4 años es del 90%. Si el tumor es de tipo anaplásico, la supervivencia a los 4 años es del 17%.
Etapa V (5% de los pacientes)
En esta etapa, el tumor afecta a ambos riñones al momento del diagnóstico.
- Nota: Para los pacientes con tumores en ambos riñones, cada riñón se clasifica por separado según las etapas I a III, basándose en qué tan extendida está la enfermedad antes de tomar una muestra.
- Pronóstico: La supervivencia a los 4 años es del 94% para aquellos cuya lesión más avanzada no supera la etapa I o II, y del 76% para aquellos cuya lesión más avanzada era la etapa III.
- Tratamiento: El tratamiento es personalizado y se basa en el tamaño y la cantidad de tumores.
Tumores anaplásicos en etapas I-IV
Los niños con tumores anaplásicos en etapa I tienen un muy buen pronóstico (80-90% de supervivencia a los cinco años). Se pueden tratar con el mismo plan que los pacientes de etapa I con tumores de tipo favorable.
Los niños con tumores anaplásicos difusos en etapas II a IV se consideran de alto riesgo. Estos tumores son más difíciles de tratar con la quimioterapia tradicional y necesitan tratamientos más intensos.
Galería de imágenes
-
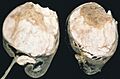
Nefroblastoma en un riñón cortado por la mitad.
-
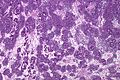
Imagen del tumor mostrando su patrón característico con túbulos, células redondas y tejido de soporte.
-
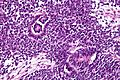
Imagen de cerca mostrando las células epiteliales (túbulos).



Véase también
 En inglés: Wilms' tumor Facts for Kids
En inglés: Wilms' tumor Facts for Kids
