
Sarcoma de Ewing para niños
Datos para niños Sarcoma de Ewing |
||
|---|---|---|
.jpg) Rayos X de un niño con sarcoma de Ewing en la tibia
|
||
| Especialidad | Oncología, oncología pediátrica | |
| Síntomas | Inflamación, rigidez y/o dolor en la localización del tumor | |
| Complicaciones | Derrame pleural, paraplejía | |
| Causas | Desconocidas | |
| Diagnóstico | Radiografía, biopsia | |
| Tratamiento | quimioterapia, radioterapia, cirugía | |
| Pronóstico | Supervivencia a los 5 años >70% | |
| Frecuencia | Incidencia de 2,93 casos por cada millón de habitantes | |
| Sinónimos | ||
| Tumor de Ewing | ||
El sarcoma de Ewing es un tipo de tumor (un bulto de células) que es dañino. Se considera una enfermedad poco común. En este tipo de tumor, las células que crecen de forma descontrolada se encuentran en los huesos o en los tejidos blandos del cuerpo. Las zonas donde aparece con más frecuencia son la pelvis, el fémur (hueso del muslo), el húmero (hueso del brazo) y las costillas.
A veces, el sarcoma de Ewing se agrupa con otros tumores que tienen una característica genética similar. Sin embargo, el sarcoma de Ewing afecta los huesos con mucha más frecuencia.
Este tumor se presenta más a menudo en hombres adolescentes. Por cada mujer que lo padece, hay aproximadamente 1.6 hombres con la enfermedad.
Aunque se clasifica como un tumor de los huesos, el sarcoma de Ewing tiene características que hacen difícil clasificarlo completamente.
Contenido
¿De dónde viene el nombre de Sarcoma de Ewing?
El nombre de este tumor viene de James Ewing (1866-1943). Él fue el primero en describir este tumor. James Ewing se dio cuenta de que esta enfermedad era diferente de otros tipos de cáncer conocidos en su época.
¿Qué tan común es el Sarcoma de Ewing?
El sarcoma de Ewing es el segundo tipo de cáncer de hueso más común en adolescentes y adultos jóvenes. La edad promedio de las personas que lo padecen es de 15 años. Representa menos del 5% de todos los tumores de tejidos blandos.
Entre 1973 y 2004, en Estados Unidos, se encontraron aproximadamente 2.93 casos por cada millón de personas. A nivel mundial, la cantidad de casos nuevos es menor de 2 por cada millón de niños al año. La mayoría de los casos ocurren entre los 10 y los 15 años de edad. Un 30% de los casos se dan en niños menores de 10 años. Otro 30% ocurre en adultos jóvenes mayores de 20 años. No se sabe qué tan común es el sarcoma de Ewing en personas mayores.
Es más común en hombres que en mujeres, con una proporción de 3 hombres por cada mujer. Además, las personas de origen caucásico (piel clara) tienen más probabilidades de padecerlo que las de origen asiático, latino, afroamericano o africano.
¿Qué causa el Sarcoma de Ewing?
El sarcoma de Ewing es causado por un cambio genético entre dos cromosomas: el 11 y el 22. Este cambio hace que dos genes, el EWS del cromosoma 22 y el FLI1 del cromosoma 11, se unan. Esta unión de genes, llamada fusión EWS/FLI, actúa como un "controlador principal" que puede hacer que las células se vuelvan cancerosas.
Existen otros cambios genéticos menos comunes que también pueden causar este tumor.
¿Cuáles son los signos y síntomas del Sarcoma de Ewing?
Este tumor puede aparecer en cualquier parte del cuerpo. Sin embargo, es más frecuente en la pelvis y en los huesos largos de los brazos y las piernas, especialmente cerca de las zonas de crecimiento. La parte central del fémur es uno de los lugares más comunes. Le siguen la tibia (hueso de la espinilla) y el húmero.
Las personas con sarcoma de Ewing suelen sentir dolor intenso en el hueso, rigidez o hinchazón en la zona del tumor. Más de la mitad de los pacientes tienen un dolor que aparece y desaparece, y que empeora por la noche.
Si el tumor está en la pelvis, puede causar dolor en la parte baja de la espalda. Si el tumor se ha extendido a otras partes del cuerpo, la persona puede tener síntomas como fiebre y pérdida de apetito. Aproximadamente el 20% de los pacientes ya tienen el tumor extendido a otras partes del cuerpo cuando se les diagnostica. De ellos, un 20% tienen problemas en los pulmones.
¿Cómo se diagnostica el Sarcoma de Ewing?

Para diagnosticar el sarcoma de Ewing, los médicos usan varias pruebas:
- Radiografías: Las radiografías comunes pueden mostrar una lesión en el hueso. A veces, la reacción del hueso alrededor del tumor se ve como capas de "piel de cebolla". Las radiografías son muy útiles para la primera evaluación.
- Resonancia Magnética (IRM): La resonancia magnética se usa para ver la extensión completa del tumor en el hueso y en los tejidos blandos cercanos. También ayuda a ver cómo el tumor se relaciona con las estructuras cercanas, como los vasos sanguíneos.
- Tomografía Axial Computarizada (TAC): La TAC también puede usarse para ver qué tan extendido está el tumor fuera del hueso, especialmente en la cabeza, la columna vertebral, las costillas y la pelvis. Tanto la TAC como la IRM pueden usarse para ver cómo responde el tumor al tratamiento.
Además de las imágenes, se realizan pruebas de laboratorio para confirmar el diagnóstico.
¿Con qué otras enfermedades se puede confundir?
Otras enfermedades pueden parecerse al sarcoma de Ewing. Algunas de ellas son:
- Infecciones en el hueso (osteomielitis).
- Otros tipos de tumores como el neuroblastoma, el rabdomiosarcoma o el linfoma.
- Otros tumores de hueso como el osteosarcoma.
- Algunas lesiones benignas (no dañinas) que afectan el hueso.
¿Cómo se trata el Sarcoma de Ewing?
El tratamiento para el sarcoma de Ewing suele incluir una combinación de diferentes métodos:
- Quimioterapia: Es el primer tratamiento que se usa. Consiste en medicamentos que ayudan a reducir el tamaño del tumor. Esto hace que sea más fácil de quitar con cirugía. Se usan diferentes combinaciones de medicamentos, como el protocolo VACA o el VAC-IE.
- Cirugía: Después de la quimioterapia, se realiza una cirugía para quitar el tumor. A veces, esto significa reemplazar una parte del hueso con material especial. En algunos casos, si el tumor es muy grande o está en un lugar difícil, puede ser necesario quitar una parte o todo el miembro afectado (amputación). Si el tumor está en la pelvis, la cirugía es menos común y se usa más la radioterapia.
- Radioterapia: Se usan radiaciones para destruir las células del tumor. Se puede usar antes de la cirugía para reducir el tamaño del tumor o después de la cirugía para eliminar cualquier célula tumoral que haya quedado. Los tratamientos que combinan cirugía con radioterapia suelen ser más efectivos.
- Rehabilitación: Después del tratamiento, es importante la rehabilitación. Esto ayuda a las personas a recuperar su autonomía y a manejar los efectos del cáncer y su tratamiento. A menudo, esto incluye la ayuda de fisioterapeutas y técnicos ortopédicos, especialmente si se necesita una prótesis después de una amputación.
Galería de imágenes
-
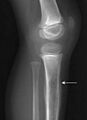
Rayos X de un niño con sarcoma de Ewing en la tibia
-
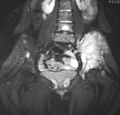
Corte de imagen por resonancia magnética mostrando el sarcoma de Ewing de la cadera izquierda (área blanca resaltada a la derecha).
Véase también
 En inglés: Ewing sarcoma Facts for Kids
En inglés: Ewing sarcoma Facts for Kids
