
Biología computacional para niños
La biología computacional es un campo de la ciencia que usa algoritmos y computadoras para entender mejor los problemas de la biología. Imagina que es como un detective que usa herramientas digitales para resolver misterios sobre los seres vivos.
Esta área de estudio se enfoca en analizar grandes cantidades de datos biológicos, crear modelos matemáticos y hacer simulaciones en la computadora. Los sistemas que estudia van desde las partes más pequeñas de las células, como las moléculas, hasta organismos completos, el sistema nervioso, y hasta cómo interactúan los seres vivos en un ecosistema.
La biología computacional combina conocimientos de muchas otras ciencias, como la química, la bioquímica, la genética, las matemáticas, la ingeniería de sistemas, la física y la estadística.
Contenido
- ¿Qué es la Biología Computacional?
- Áreas de Estudio
- Bioinformática: Herramientas para la Biología
- Biomodelado Computacional: Creando Modelos de la Vida
- Biología de Sistemas: El Panorama Completo
- Biología Evolutiva: Entendiendo el Pasado
- Genómica Computacional: El Libro de la Vida
- Neurociencia Computacional: El Cerebro y las Computadoras
- Farmacología: Diseñando Medicamentos
- Programas y Herramientas
- Galería de imágenes
- Véase también
¿Qué es la Biología Computacional?
A veces, la biología computacional se confunde con la Bioinformática, o se piensa que son muy parecidas. Una organización importante llamada NIH (Institutos Nacionales de Salud) las define como campos distintos, aunque se superponen un poco.
Según esta definición, la bioinformática se encarga más de crear herramientas y programas de computadora para organizar, analizar y procesar los datos biológicos. La biología computacional, en cambio, usa esas herramientas para estudiar los sistemas biológicos completos, como una célula o un órgano, usando la computadora.
Áreas de Estudio
Bioinformática: Herramientas para la Biología
La bioinformática es una parte de la biología computacional que se dedica a investigar, desarrollar y usar programas de computadora. Estos programas sirven para obtener, guardar, organizar, analizar y mostrar datos biológicos.
Es un campo que mezcla muchas disciplinas. Utiliza métodos y herramientas de minería de datos (para encontrar patrones en grandes cantidades de información), reconocimiento de patrones (para identificar cosas que se repiten), aprendizaje automático (para que las computadoras aprendan de los datos) y procesamiento digital de imágenes. Todo esto se usa para resolver preguntas biológicas, como:
- Comparar secuencias de ADN o proteínas.
- Predecir dónde están los genes.
- Comparar los genomas (todo el ADN) de diferentes especies.
- Predecir cómo se doblan las proteínas.
- Modelar cómo interactúan las moléculas.
.png)
Los temas más importantes en bioinformática son el análisis de secuencias de ADN y ARN, y el estudio de cómo se "encienden" o "apagan" los genes (su regulación).
El análisis de secuencias busca identificar partes específicas en el ADN, encontrar patrones o secuencias que se repiten, y reconocer características genéticas. Por ejemplo, busca genes que producen proteínas o ARN, o lugares donde se unen ciertas moléculas para controlar los genes. Todo esto es importante para entender cómo funcionan las células y los organismos.
Gracias a la bioinformática, se han creado grandes bases de datos como RefSeq y GenBank, que guardan muchísimas secuencias biológicas. También se han desarrollado herramientas computacionales como BLAST y ClustalW para comparar secuencias, o GLIMMER y GENSCAN para encontrar genes.
El análisis de la expresión génica se refiere a medir cuántas copias de ARN o proteínas hay en una célula. La bioinformática es clave para desarrollar herramientas que aseguren que los datos obtenidos sean de buena calidad y confiables. A menudo, los experimentos generan mucha información no deseada, y la bioinformática ayuda a "limpiar" esos datos para obtener resultados claros.
Biomodelado Computacional: Creando Modelos de la Vida
Esta área se enfoca en crear modelos de sistemas biológicos usando computadoras. Es como construir una versión virtual de una célula o un órgano para ver cómo funciona.
Biología de Sistemas: El Panorama Completo

La biología de sistemas estudia los sistemas biológicos completos, desde las moléculas hasta poblaciones enteras. Lo hace creando modelos matemáticos de sus partes, cómo interactúan entre sí y las propiedades que surgen de esas interacciones.
A diferencia de estudiar las partes por separado, la biología de sistemas busca entender el "panorama completo" de los procesos biológicos. Los sistemas más estudiados son las rutas metabólicas (como la glucólisis, que es cómo las células obtienen energía del azúcar) y las vías de señalización celular (cómo las células se comunican y responden a su entorno).
Las ciencias "ómicas" (como la metabolómica, que estudia todos los metabolitos; la proteómica, que estudia todas las proteínas; o la genómica, que estudia todos los genes) están muy relacionadas con la biología de sistemas. Estas ciencias generan muchísimos datos que la biología de sistemas usa para construir sus modelos.
Un campo muy cercano es la biología sintética. Esta busca mejorar sistemas biológicos que ya existen en la naturaleza (por ejemplo, dándole nuevas funciones a una enzima) o diseñar y construir nuevos sistemas biológicos usando ingeniería genética. Ambos campos se ayudan mutuamente.
Biología Evolutiva: Entendiendo el Pasado
Las herramientas computacionales y estadísticas son muy útiles para estudiar cómo se relacionan las moléculas (como las proteínas) o los individuos a lo largo de la evolución. Permiten reconstruir el "árbol genealógico" de la vida.
Genómica Computacional: El Libro de la Vida

La genómica computacional se dedica a estudiar las secuencias completas de los genomas (el ADN o ARN de un organismo) usando herramientas de computadora y estadística. Dos de los estudios más comunes son la comparación de secuencias y la secuenciación del ADN.
Para comparar secuencias, se usan algoritmos como BLAST para ver qué tan parecidas son dos o más secuencias.
Para la secuenciación del ADN, que es leer el "código" genético, existen muchos métodos. Los avances en estos métodos han hecho que se necesiten herramientas computacionales cada vez más potentes para manejar la enorme cantidad de datos que se generan. Estas herramientas ayudan a identificar las "letras" del ADN, a comparar la secuencia obtenida con un genoma de referencia y a encontrar pequeñas diferencias genéticas.
Un gran ejemplo de genómica computacional a nivel mundial fue el Proyecto Genoma Humano. En 2003, se logró secuenciar por primera vez casi todo el genoma humano con una precisión muy alta. Después de este éxito, se han realizado muchos otros proyectos para secuenciar genomas, como el Proyecto 1000 genomas, que estudia la diversidad del genoma humano en todo el mundo, o el Proyecto Earth BioGenome, que busca secuenciar todos los organismos eucariotas de la Tierra para ayudar a conservar la biodiversidad.
Neurociencia Computacional: El Cerebro y las Computadoras
Esta área usa modelos y simulaciones computacionales para entender cómo funciona el cerebro y el sistema nervioso.
Farmacología: Diseñando Medicamentos
En Farmacología, la biología computacional ayuda a entender cómo interactúan los medicamentos con el cuerpo y a diseñar nuevas medicinas de manera más eficiente.
Programas y Herramientas
Los biólogos computacionales usan muchas herramientas en sus computadoras. Pueden ser programas sencillos que se ejecutan con comandos o programas más complejos con interfaces gráficas. Es común que ellos mismos escriban sus propios programas, desde pequeños "scripts" para analizar datos hasta programas muy grandes.
Programas de Código Abierto
Los programas de Código abierto (y Software libre) son muy importantes en la ciencia. Permiten que cualquiera pueda ver, corregir y mejorar el código de un programa. La revista PLOS Computational Biology menciona cuatro razones principales para usar código abierto en la ciencia:
- Reproducibilidad: Permite que otros investigadores usen exactamente los mismos métodos para analizar datos, lo que hace que los resultados sean más confiables.
- Desarrollo más rápido: Los científicos pueden usar código que ya existe y adaptarlo a sus necesidades, en lugar de empezar desde cero.
- Mayor calidad: Al ser el código accesible para todos, es más fácil encontrar y corregir errores.
- Disponibilidad a largo plazo: El código abierto no depende de una sola empresa o patente, lo que ayuda a que esté disponible por mucho tiempo y se difunda ampliamente.
Galería de imágenes
-

Ejemplo de alineamiento múltiple de secuencias de la proteína ZNF226, procedentes de 20 especies diferentes en un estudio de búsqueda de patrones conservados evolutivamente.
-
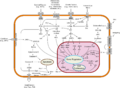
Ejemplos de rutas de transducción de señales a nivel intracelular, que en conjunto pueden suponer un sistema biológico. Diferentes moléculas (ej.: hormonas, citocinas, factores de crecimiento etc.) pueden actuar en sus respectivos receptores diana en la célula de destino. Esto puede generar una serie de reacciones en cadena entre diferentes proteínas intracelulares, provocando una respuesta (ej.: activación/desactivación de la expresión génica, promover la proliferación celular o la apoptosis.
-

Ideograma del cromosoma X del genoma humano (extraído del NCBI)
Véase también
 En inglés: Computational biology Facts for Kids
En inglés: Computational biology Facts for Kids
