
Síndromes de Ehlers-Danlos para niños
Datos para niños Síndromes de Ehlers-Danlos |
||
|---|---|---|
.png) Individuo con SED tipo clásico mostrando hiperelasticidad de la piel
|
||
| Especialidad | Genética médica, reumatología | |
| Síntomas | Hiperlaxitud, hiperelasticidad de la piel, trastornos de la formación de tejido cicatricial | |
| Complicaciones | Disección aórtica, luxación articular, artrosis | |
| Duración | Crónica | |
| Causas | Genéticas | |
| Factores de riesgo | Historial familiar | |
| Diagnóstico | Basado en síntomas, examen genético, biopsia de la piel | |
| Diagnóstico diferencial | Osteogénesis imperfecta, Síndrome de Marfan, Síndrome de Loeys-Dietz, Cutis laxa, fibromialgia, síndrome de fatiga crónica | |
| Tratamiento | Orientado a evitar complicaciones, evitar deportes de impacto, realización de ejercicio físico de carga ligera o moderada | |
| Pronóstico | Variable según el subtipo | |
| Frecuencia | Incidencia de 1 por cada 2500—5000 | |
| Sinónimos | ||
| Enfermedad de Danlos; Enfermedad de Danlos, Ehlers; Síndrome de Danlos-Ehlers; Enfermedad de Ehlers-Danlos; Enfermedad de Ehlers-Danlos; Distrofia mesodérmica congénita; EDS; Síndrome de Ehler-Danlos; Enfermedad de Ehlers-Danlos; Síndromes de Ehlers-Danlos; Piel elástica; Fibrodisplasia elástica generalizada; Displasia hereditaria del colágeno; Síndrome de Meekeren-Ehlers-Danlos; Piel elástica; Síndrome de Ehlers-Danlos | ||
Los síndromes de Ehlers-Danlos (SED) son un grupo de trastornos genéticos que se heredan y afectan a las personas. Son causados por un problema en la forma en que el cuerpo produce el colágeno. El colágeno es una proteína muy importante que da fuerza y elasticidad a muchas partes del cuerpo.
Generalmente, los SED se caracterizan por tener hipermovilidad en las articulaciones (lo que significa que se estiran más de lo normal), piel muy elástica y tejidos frágiles. La gravedad de estos síndromes puede variar mucho, desde muy leve hasta causar problemas de salud serios. Aunque no hay una cura, existen tratamientos para ayudar a manejar los síntomas y prevenir complicaciones.
Contenido
¿Qué son los Síndromes de Ehlers-Danlos?
Los Síndromes de Ehlers-Danlos son afecciones que se heredan de los padres a los hijos. Esto significa que son causados por cambios en los genes. Estos cambios afectan la producción o el funcionamiento del colágeno. El colágeno es como el "pegamento" que mantiene unidos los tejidos del cuerpo, como la piel, los huesos, los vasos sanguíneos y los órganos. Cuando el colágeno no funciona bien, estos tejidos pueden volverse débiles o demasiado elásticos.
¿De dónde viene el nombre?
El nombre de estos síndromes viene de dos médicos que los describieron por primera vez. El doctor Edvard Ehlers, de Dinamarca, los describió en 1901. Él notó que las personas tenían la piel muy elástica, articulaciones que se estiraban mucho y moretones con facilidad. Luego, en 1908, el doctor Henri-Alexandre Danlos, de Francia, también observó estos síntomas en un paciente.
¿Cuántas personas tienen SED?
Se calcula que los síndromes de Ehlers-Danlos afectan a entre 1 de cada 2500 y 1 de cada 5000 bebés que nacen. Esto incluye todos los tipos de SED.
El tipo más común es el de hipermovilidad, que afecta a entre 1 de cada 10 000 y 1 de cada 15 000 personas. El tipo clásico es un poco menos común, afectando a 1 de cada 10 000 a 20 000 personas. Otros tipos son mucho más raros. Por ejemplo, el tipo cifoescoliótico se ha reportado en menos de 60 pacientes en todo el mundo. Es posible que haya más personas con SED de lo que se sabe, porque los casos más leves a veces no se diagnostican.
¿Cómo afectan los SED al cuerpo?

Los síntomas de los SED varían mucho de una persona a otra, dependiendo del tipo específico y de cómo el colágeno está afectado. Sin embargo, todos los síntomas se relacionan con la falta o el mal funcionamiento del colágeno.
Algunos síntomas comunes incluyen:
- Articulaciones muy flexibles: Las articulaciones pueden estirarse más de lo normal y ser inestables. Esto puede causar que se salgan de su lugar (dislocaciones o subluxaciones) y provocar dolor. Esto ocurre porque los ligamentos, que conectan los huesos, son demasiado elásticos.
- Piel elástica y frágil: La piel puede estirarse mucho y ser suave. También puede ser más propensa a moretones o a rasgarse con facilidad, y las heridas pueden tardar más en sanar.
- Cansancio: Muchas personas con SED sienten mucho cansancio, especialmente al hacer ejercicio.
- Problemas circulatorios: Pueden presentarse alteraciones en la circulación de la sangre.
En los tipos más graves, como el SED vascular, puede haber complicaciones serias en los vasos sanguíneos o en los órganos internos. Esto requiere un cuidado médico muy atento.
Otros problemas de salud relacionados
Las personas con síndromes de Ehlers-Danlos a veces tienen otras condiciones de salud que pueden hacer que los síntomas sean más difíciles de manejar. Algunas de estas condiciones incluyen:
- Enfermedad celíaca: Una condición donde el cuerpo no puede tolerar el gluten.
- Fibromialgia: Una condición que causa dolor generalizado en el cuerpo.
- Síndrome de fatiga crónica: Una condición que causa cansancio extremo y persistente.
- Deficiencia de vitamina D.
- Malformación de Chiari: Un problema en la parte inferior del cerebro.
Tipos de Síndromes de Ehlers-Danlos
Actualmente, los expertos reconocen 13 tipos diferentes de síndromes de Ehlers-Danlos. Para la mayoría de estos tipos, se han identificado los cambios genéticos específicos que los causan. Esto es muy útil para hacer un diagnóstico preciso, ya que los síntomas pueden ser muy parecidos entre los diferentes tipos.
Aquí te presentamos algunos de los tipos principales:
| Nombre del Tipo | Características Principales | Gen(es) Asociados |
| Clásico | Piel suave, muy elástica y frágil que se lastima fácilmente. Las heridas sanan lentamente y pueden dejar cicatrices finas. Las articulaciones son muy flexibles. | HGNC COL5A1 , HGNC COL5A2 , HGNC COL1A1 |
| Hipermovilidad | Es el tipo más común. Las articulaciones son muy inestables y laxas, lo que causa dislocaciones frecuentes y dolor. La piel puede ser suave y moderadamente elástica. | Se desconoce (el diagnóstico se basa en los síntomas) |
| Vascular | Es un tipo más serio. La piel es muy delicada y transparente, y los vasos sanguíneos son frágiles. Puede haber riesgo de problemas graves en arterias o en órganos internos. La flexibilidad de las articulaciones suele limitarse a los dedos. | HGNC COL3A1 |
| Cifoscoliosis | Causa una curvatura progresiva de la columna vertebral (escoliosis) y debilidad muscular. Los ojos pueden tener la parte blanca (esclerótica) frágil. Es muy raro. | HGNC PLOD1 |
| Artrocalasia | Las articulaciones son extremadamente laxas e inestables, especialmente las caderas, lo que puede llevar a problemas articulares tempranos. La piel es elástica y frágil. Es muy raro. | HGNC COL1A1 , HGNC COL1A2 |
| Dermatosparaxis | La piel es extremadamente frágil y pierde su elasticidad. Las articulaciones también pueden ser inestables. Es un tipo muy raro. | HGNC ADAMTS2 |
Es importante saber que los síntomas de los diferentes tipos de SED a menudo se superponen. Esto significa que una persona puede tener síntomas de varios tipos, lo que a veces hace que el diagnóstico sea un desafío.
¿Qué causa los SED?
Los síndromes de Ehlers-Danlos son causados por cambios (mutaciones) en ciertos genes. Estos genes son responsables de producir o procesar el colágeno o proteínas que interactúan con él.
Algunos de los genes que pueden estar involucrados son:
- Proteínas fibrosas: COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, y TNXB.
- Enzimas: ADAMTS2 y PLOD1.
Cuando hay un problema en estos genes, el colágeno no se forma correctamente. Esto debilita el tejido conectivo en todo el cuerpo, lo que lleva a los síntomas que se observan en los síndromes de Ehlers-Danlos.
¿Cómo se heredan los SED?
La forma en que se heredan los SED depende del tipo específico. La mayoría de los tipos se heredan de forma autosómica dominante. Esto significa que si una persona hereda solo una copia del gen alterado de uno de sus padres, desarrollará el síndrome.
Una minoría de los tipos se heredan de forma autosómica recesiva. En este caso, una persona necesita heredar dos copias del gen alterado (una de cada padre) para que los síntomas se manifiesten.
SED en animales
Se han observado condiciones similares a los síndromes de Ehlers-Danlos en algunos animales, como gatos (incluyendo los de la raza Himalaya), y ciertas razas de ganado vacuno. También se ha visto de forma ocasional en perros domésticos.
Galería de imágenes
-
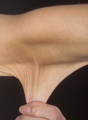
Individuo con SED tipo clásico mostrando hiperelasticidad de la piel
-

Paciente con síndrome de Ehlers-Danlos que muestra articulaciones hiperelásticas.
Véase también
 En inglés: Ehlers–Danlos syndrome Facts for Kids
En inglés: Ehlers–Danlos syndrome Facts for Kids
