
Proyecto del genoma neandertal para niños

Datos para niños Genética humana |
||
|---|---|---|
| Subtemas |
|
|
El proyecto del genoma del neandertal fue una investigación muy importante que comenzó en julio de 2006. Fue anunciada por el Instituto Max Planck de Antropología Evolutiva en Alemania y la empresa 454 Life Sciences de Estados Unidos. Su objetivo principal era estudiar el ADN de los neandertales.
Los resultados de este proyecto se publicaron en la revista Science en mayo de 2010. Allí se presentó un primer borrador del genoma del neandertal. Este estudio analizó una enorme cantidad de información genética y descubrió algo sorprendente: hubo un intercambio de genes entre los neandertales y los primeros humanos modernos. Esto significa que una parte del genoma neandertal todavía se encuentra en las personas de hoy que viven fuera de África. En 2013, el equipo de Svante Pääbo confirmó esta mezcla de genes al secuenciar el genoma completo del neandertal.
Contenido
¿Qué se descubrió sobre el genoma neandertal?
El genoma del neandertal, que es como el "manual de instrucciones" genético de esta especie, tiene un tamaño muy parecido al nuestro, con unos 3.200 millones de pares de bases. Las primeras investigaciones mostraron que el ADN de los neandertales y el de los humanos modernos son casi idénticos en un 99.7%. Para que te hagas una idea, la diferencia entre los humanos modernos y los chimpancés es mayor, alrededor del 98.8% o incluso el 94% en otros estudios.
¿De dónde se obtuvo el ADN neandertal?
Para conseguir el ADN neandertal, los científicos extrajeron pequeñas muestras de huesos, como el fémur. Usaron huesos de tres hembras neandertales que vivieron hace unos 38.000 años, encontrados en la cueva de Vindija en Croacia. También se usaron muestras de España, Rusia y Alemania. Solo se necesitó medio gramo de hueso para obtener la secuencia genética.
El proyecto tuvo varios desafíos. Uno de los mayores problemas fue la contaminación del material. Las muestras podían contaminarse con bacterias o con el ADN de las personas que las manipularon en los lugares de excavación o en el laboratorio. Para evitar esto, se elegía la muestra con menos contaminación. Además, como el ADN era muy antiguo, era fácil que se introdujeran errores durante el proceso de secuenciación, y los científicos tuvieron que corregirlos cuidadosamente.
El homínido de Denisova y su relación
En 2010, se analizó el ADN mitocondrial de otro grupo de humanos antiguos, el homínido de Denisova, encontrado en las cuevas de Denisova en Siberia. Este análisis mostró que el ADN de los denisovanos era diferente al de los humanos modernos y al de los neandertales. El equipo de Svante Pääbo en el Instituto Max Planck lideró este estudio y confirmó que los denisovanos eran una especie distinta. Se cree que los denisovanos y los neandertales tuvieron un ancestro común hace unos 650.000 años, y que los denisovanos y los humanos modernos compartieron un ancestro hace unos 800.000 años.
Más tarde, en la misma cueva de Denisova, se encontró un hueso del pie de un neandertal adulto. Este hueso permitió obtener una secuencia completa y de muy buena calidad del genoma neandertal. A este individuo se le llamó el "Neandertal de Altái".
¿Cómo avanzó la investigación del genoma neandertal?
En 2006, dos equipos de científicos publicaron sus primeros resultados sobre el ADN neandertal:
- El equipo de Richard Green publicó en la revista Nature.
- El equipo de Noonan publicó en la revista Science.
Al principio, hubo algunas dudas sobre si realmente había habido una mezcla de genes entre neandertales y humanos modernos. Sin embargo, se encontró que el gen FoxP2, relacionado con el lenguaje, era igual en neandertales y humanos modernos. Esto sugiere que los neandertales podrían haber tenido algunas habilidades básicas de lenguaje.
Técnicas de secuenciación de ADN
El equipo de Richard Green usó una técnica nueva en ese momento, desarrollada por 454 Life Sciences, que permitía obtener muchas secuencias cortas de ADN. Esta técnica era muy útil, pero destruía la muestra original.
El equipo de Noonan, dirigido por Edward Rubin, usó una técnica diferente. Insertaron el ADN neandertal en bacterias para que estas hicieran muchas copias de los fragmentos. Esto les permitió estudiar el genoma de una manera diferente.
Aunque usaron métodos distintos, los resultados de ambos equipos fueron muy parecidos. Al principio, uno de los equipos sugirió que pudo haber mezcla de genes, mientras que el otro no encontró pruebas claras. Ambos concluyeron que necesitaban más datos para estar seguros.
¿Cuándo se separaron neandertales y humanos modernos?
Basándose en sus estudios, el equipo de Noonan calculó que el ancestro común más reciente de los neandertales y los humanos modernos vivió hace unos 706.000 años. La separación de las poblaciones ancestrales de neandertales y humanos modernos habría ocurrido hace unos 370.000 años.
Investigaciones anteriores, como la de Svante Pääbo en 1997, habían estimado que la separación de los linajes fue hace unos 500.000 años. El equipo de Green calculó una antigüedad de 516.000 años para el ancestro común.
En febrero de 2009, el equipo de Svante Pääbo anunció que había completado el primer borrador del genoma neandertal. Aunque al principio se pensó que los neandertales no habían dejado rastro genético en los humanos modernos, estudios posteriores demostraron lo contrario. Pääbo también mencionó que, a partir de ADN fósil, es imposible crear un neandertal mediante clonación.
En mayo de 2010, el proyecto publicó un borrador más completo del genoma neandertal. Este estudio confirmó que entre el 1% y el 4% del ADN de los humanos modernos fuera de África proviene de los neandertales. Esto sugiere que el cruce entre ambas especies pudo haber ocurrido en Oriente Medio antes de que los humanos modernos se extendieran por Europa.
El genoma completo del neandertal
En enero de 2014, Svante Pääbo publicó en la revista Nature las conclusiones del genoma completo del neandertal. Este estudio usó el ADN de la falange del pie del "Neandertal de Altái". Los análisis genéticos confirmaron que este individuo estaba muy relacionado con otros neandertales, como el niño encontrado en la cueva Mezmaiskaya.

El estudio del genoma de este neandertal también mostró que sus padres estaban muy emparentados, lo que indica un alto grado de endogamia en la población neandertal. Esto sugiere que las poblaciones neandertales no eran muy grandes y se expandieron en grupos pequeños. A diferencia de los humanos modernos, cuyas poblaciones ancestrales lograron recuperarse después de una reducción de tamaño hace más de un millón de años, las poblaciones de Altái y Denisova continuaron disminuyendo.
Los cálculos más precisos, usando este genoma completo, indican que los humanos modernos se separaron de los neandertales y denisovanos hace entre 553.000 y 589.000 años. Los neandertales y los denisovanos se separaron entre sí hace unos 381.000 años.
¿Cuánta herencia genética tenemos de neandertales y denisovanos?
Hoy en día, las personas que viven fuera de África tienen entre un 1.5% y un 2.1% de su genoma heredado de los neandertales. Los denisovanos también dejaron su huella genética, especialmente en las poblaciones de Oceanía (entre 3% y 6%) y Asia (0.2%). Esto demuestra que hubo un intercambio genético entre estas especies.
También se descubrió que los denisovanos se cruzaron con un cuarto grupo de humanos antiguos que vivía en Eurasia. Se cree que este grupo podría ser el Homo erectus. Además, hubo un pequeño intercambio genético entre neandertales y denisovanos.
Este proyecto ha creado un catálogo de diferencias genéticas entre los humanos modernos, los neandertales, los denisovanos y los simios. Se encontraron pocas diferencias importantes, afectando a solo 96 cambios en 87 proteínas. Estos 87 genes específicos en los humanos modernos, que son diferentes de los de neandertales y denisovanos, podrían darnos pistas sobre las características que nos distinguen y que permitieron a los humanos modernos expandirse y desarrollarse, mientras que otras especies se extinguieron.
Véase también
 En inglés: Neanderthal genome project Facts for Kids
En inglés: Neanderthal genome project Facts for Kids
- Flujo genético
- Híbrido (biología)
- Homo neanderthalensis
- Hombre de Cro-Magnon
- Homo sapiens
- Proyecto Genoma Humano
- Neanderthal Man: In Search of Lost Genomes
Galería de imágenes
-
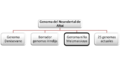
Comparación del Genoma del Neandertal de Altái con genomas de otros homínidos por Prüfer et al., 2014. Enmarcado el homínido con mayor relación
