
Enfermedad de almacenamiento de glucógeno tipo II para niños
Datos para niños Enfermedad de Pompe. |
||
|---|---|---|
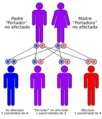
 La enfermedad de Pompe tiene una patrón de herencia autosómico recesivo
|
||
| Especialidad | endocrinología | |
| Sinónimos | ||
|
||
La enfermedad de Pompe, también conocida como glucogenosis tipo II, es una condición poco común que se hereda. Ocurre cuando el cuerpo no produce suficiente cantidad de una enzima especial llamada alfa-glucosidasa ácida. Esta enzima es como una "tijera" que ayuda a descomponer el glucógeno (una forma de azúcar que el cuerpo almacena para obtener energía) dentro de unas partes de las células llamadas lisosomas.
Cuando la enzima no funciona bien, el glucógeno se acumula en los lisosomas, especialmente en los músculos. Esto puede hacer que los músculos no funcionen correctamente. En los niños, esta acumulación puede afectar el músculo cardíaco, haciendo que el corazón crezca más de lo normal.
Contenido
¿Qué tan común es la enfermedad de Pompe?
Se calcula que la enfermedad de Pompe afecta a 1 de cada 18.698 bebés que nacen. Esta condición se presenta en personas de todas las partes del mundo. Como es una enfermedad autosómica recesiva, afecta por igual a hombres y mujeres.
En los países con más recursos, se estima que hay entre 5.000 y 10.000 personas viviendo con esta enfermedad. También se han encontrado casos en diferentes animales, como peces, aves y mamíferos.
En España, la enfermedad es más frecuente en las regiones de Andalucía, Madrid y Murcia. La provincia de Jaén, en particular, parece tener más casos, especialmente de las formas más serias de la enfermedad.
¿Qué causa la enfermedad de Pompe?
La enfermedad de Pompe es un problema congénito (presente desde el nacimiento) relacionado con cómo el cuerpo usa el glucógeno. La causa es un cambio en un gen específico, llamado GAA. Este gen es el encargado de dar las instrucciones para producir la enzima alfa-(1,4)-glucosidasa en los lisosomas.
El gen GAA se encuentra en el cromosoma 17. Dependiendo del tipo de cambio en el gen, la enzima puede ser totalmente deficiente o solo funcionar un poco. Esta deficiencia afecta a todas las células del cuerpo, pero sus efectos son más notables en las células musculares. Allí, el glucógeno se acumula en los lisosomas, lo que interfiere con el funcionamiento normal de las células y las daña. Se han descubierto cerca de 200 cambios diferentes en el gen GAA.
¿Cuáles son los tipos de enfermedad de Pompe?
La enfermedad de Pompe se presenta en tres tipos principales, que se diferencian por la edad en que aparecen los síntomas y la rapidez con la que avanza la enfermedad. Esto depende de cuánta actividad enzimática tenga el paciente:
- Tipo infantil: Los síntomas aparecen muy temprano, a menudo en los primeros meses de vida. La actividad de la enzima es muy baja (menos del 1% de lo normal).
- Tipo juvenil: Los síntomas aparecen más tarde, en la infancia o adolescencia. La actividad de la enzima está entre el 1% y el 10% de lo normal.
- Tipo adulta: Los síntomas suelen aparecer en la edad adulta. La actividad de la enzima es un poco mayor, entre el 10% y el 20% de lo normal.
¿Cómo se trata la enfermedad de Pompe?
El tratamiento de la enfermedad de Pompe busca manejar los síntomas y mejorar la calidad de vida. Las complicaciones que afectan el corazón y la respiración se tratan según sea necesario. La terapia física y terapia ocupacional pueden ser de gran ayuda para algunos pacientes.
Cambios en la alimentación pueden ofrecer mejoras temporales, pero no detienen el avance de la enfermedad. El asesoramiento genético es importante para las familias, ya que les da información sobre el riesgo de que la enfermedad se presente en futuros embarazos.
Terapia de reemplazo enzimático
Existe un tratamiento específico llamado terapia de reemplazo enzimático. En 2006, se aprobó un medicamento llamado Myozyme (alglucosidasa alfa). Este medicamento es una versión artificial de la enzima que falta en los pacientes con Pompe. Se produce usando células especiales.
Myozyme se administra a través de una infusión en la vena. Se ha demostrado que este tratamiento ayuda a prolongar la vida de los pacientes, especialmente si se diagnostica y se empieza a tratar la enfermedad a tiempo. Algunos efectos secundarios pueden incluir fiebre o erupciones en la piel, pero suelen ser manejables.
En 2010, se aprobó otro medicamento similar, Lumizyme, para tratar la enfermedad de Pompe en sus formas de aparición tardía.
Véase también
 En inglés: Glycogen storage disease type II Facts for Kids
En inglés: Glycogen storage disease type II Facts for Kids
- Glucogenosis tipo I o Enfermedad de Von Gierke
- Glucogenosis tipo III o Enfermedad de Cori
- Glucogenosis tipo V o Enfermedad de McArdle
- Glucogenosis tipo VII o Enfermedad de Tarui
- Medidas extraordinarias (película)
Galería de imágenes
-
La enfermedad de Pompe tiene una patrón de herencia autosómico recesivo
