
Reacción aldólica para niños
La reacción aldólica es un proceso químico muy importante en la química orgánica. Imagina que es como un "pegamento" especial que permite unir dos moléculas más pequeñas para formar una más grande. En esta reacción, una parte de una molécula llamada cetona (o a veces un aldehído) se une a otra molécula de aldehído.
El resultado de esta unión es una nueva molécula que a menudo se llama "aldol". El nombre "aldol" viene de "aldehído" y "alcohol", porque la molécula resultante tiene características de ambos. Esta estructura de aldol es muy común en la naturaleza y se encuentra en muchas moléculas de origen natural y en medicamentos.
A veces, después de que se forma el aldol, la molécula pierde una pequeña molécula de agua. Cuando esto sucede, la reacción se llama condensación aldólica.
La reacción aldólica fue descubierta por dos científicos, Charles-Adolphe Wurtz y Aleksandr Borodín, en el año 1872. Borodín notó que el acetaldehído podía unirse a sí mismo bajo ciertas condiciones.
Esta reacción se usa mucho en la industria para fabricar productos químicos importantes, como el pentaeritritol, y también en la industria farmacéutica para crear medicamentos con alta pureza. Por ejemplo, una empresa farmacéutica usó dos reacciones aldólicas para fabricar un medicamento para el colesterol llamado Lipitor.
Plantilla:Ficha de reacción química
La estructura de aldol es muy importante en un grupo de productos naturales llamados policétidos. De estos policétidos se derivan muchos medicamentos, como algunos que ayudan al sistema inmune, antibióticos (como las tetraciclinas) y agentes que combaten los hongos. Gracias a la investigación en la reacción aldólica, ahora es posible crear muchos de estos policétidos en el laboratorio, lo cual es muy útil porque en la naturaleza se encuentran en cantidades muy pequeñas.
En la bioquímica, la reacción aldólica es un paso clave en un proceso llamado glicólisis, que es fundamental para que las células obtengan energía. En este proceso, la reacción es ayudada por unas enzimas especiales llamadas aldolasas.
Contenido
¿Por qué es tan útil la Reacción Aldólica?
La reacción aldólica es muy valiosa en la síntesis orgánica (la creación de moléculas en el laboratorio) porque puede formar dos nuevos "centros estereogénicos". Imagina que estos centros son como puntos en la molécula donde los átomos pueden organizarse de diferentes maneras, como si fueran la mano izquierda y la mano derecha.
Los métodos modernos permiten controlar cómo se organizan estos centros. Esto es muy importante al fabricar medicamentos, porque moléculas con la misma estructura pero con una organización diferente de estos centros pueden tener efectos biológicos muy distintos en el cuerpo.
Se pueden usar diferentes tipos de moléculas para iniciar la reacción aldólica, incluyendo enoles, enolatos y éteres de enol de cetonas y aldehídos. Generalmente, la molécula que es atacada es un aldehído. Cuando las dos moléculas que reaccionan son diferentes, se le llama reacción aldólica cruzada. Si reaccionan dos moléculas iguales, se llama dimerización aldólica.

¿Cómo Funciona la Reacción Aldólica?
La reacción aldólica puede ocurrir de dos maneras principales, como si tuviera dos caminos diferentes.
Mecanismo Básico: Enoles y Enolatos
En el "mecanismo enólico", las moléculas como los aldehídos y las cetonas pueden transformarse en "enoles". Los enoles tienen una parte que puede atacar a otras moléculas. Si hay un ácido presente, este ácido también ayuda a que la otra molécula de aldehído sea más fácil de atacar. Después del ataque, se forma el aldol. A menudo, este aldol pierde agua para formar una molécula insaturada.
En el "mecanismo del enolato", las moléculas de aldehído o cetona pueden perder un átomo de hidrógeno para formar "enolatos". Los enolatos son mucho más reactivos que los enoles y pueden atacar directamente a otras moléculas. El resultado es el mismo: se forma el aldol, que luego puede perder agua.

Si las condiciones de la reacción son muy fuertes (por ejemplo, con bases muy potentes y altas temperaturas), es más probable que se forme el producto de condensación (el que pierde agua). Pero si se usan condiciones más suaves o bajas temperaturas, se puede obtener el aldol sin que pierda agua.
El Modelo de Zimmerman-Traxler
En 1957, unos científicos propusieron un modelo para entender cómo se organizan las moléculas durante la reacción aldólica. Este modelo, llamado de Zimmerman-Traxler, sugiere que las moléculas se unen a través de una estructura temporal con forma de silla. Esto ayuda a predecir cómo se organizarán los nuevos centros estereogénicos en el producto final.
Controlando la Reacción Aldólica
A veces, cuando se mezclan dos cetonas diferentes, la reacción aldólica puede producir una mezcla de hasta cuatro productos distintos. Esto es un problema si solo queremos obtener uno de ellos. Por eso, es importante "controlar" la reacción.
Control por Acidez
Si una de las moléculas es mucho más "ácida" que la otra, el control puede ser automático. La base de la reacción quitará el hidrógeno más ácido, formando un enolato solo de esa molécula. Esto funciona mejor si solo una de las moléculas tiene hidrógenos ácidos que puedan formar un enolato. Por ejemplo, si se mezcla benzaldehído con malonato de dietilo, solo se formará un producto, porque el malonato es mucho más ácido y el benzaldehído no tiene hidrógenos ácidos en la posición correcta.

Control por Orden de Adición
Otra forma común de controlar la reacción es formar primero el enolato de una de las moléculas y luego añadir lentamente la otra. Esto se hace a bajas temperaturas para asegurar que la reacción vaya en la dirección deseada y no se revierta. Por ejemplo, se puede formar el enolato de una cetona a -78 °C y luego añadir lentamente un aldehído.
Enolatos: Formación y Geometría
Los enolatos son muy importantes en la reacción aldólica. Se pueden formar usando bases fuertes o ácidos de Lewis. Se ha investigado mucho cómo se forman los enolatos y cómo se puede controlar su forma.
Para las cetonas, la mayoría de las condiciones de formación de enolatos producen los enolatos de tipo "Z". Para los ésteres, la mayoría de las condiciones producen los enolatos de tipo "E". Estas letras (Z y E) se refieren a cómo están organizados los átomos alrededor de un doble enlace en la molécula.
Estereoselectividad en la Reacción Aldólica
Como mencionamos, la reacción aldólica es especial porque crea dos nuevos centros estereogénicos. Esto significa que el producto puede tener diferentes formas en el espacio. Los científicos usan términos como "syn" y "anti" para describir cómo se organizan estos centros.

La forma del enolato (E o Z) y el tipo de catión metálico que se usa pueden influir mucho en la estereoselectividad, es decir, en qué forma espacial del producto se obtiene más. El boro, por ejemplo, se usa a menudo porque sus enlaces son más cortos y esto ayuda a "fortalecer" la estructura temporal de la reacción, lo que mejora el control.
También, si las moléculas iniciales ya tienen un centro estereogénico, esto puede influir en el resultado de la reacción, un efecto llamado "estereocontrol basado en el sustrato".
Química Aldólica Moderna
La química moderna busca crear compuestos en una forma "enantiopura", lo que significa que solo se obtiene una de las dos formas "mano izquierda" o "mano derecha" de la molécula. Como la reacción aldólica puede crear hasta cuatro formas diferentes, se han desarrollado muchos métodos para controlar esto.
El Método de la Oxazolidinona de Evans
Un método muy usado es el de la oxazolidinona de acilo de Evans, desarrollado por David A. Evans. Este método funciona añadiendo una "ayuda quiral" (la oxazolidinona) a la molécula. Esta ayuda tiene una forma espacial definida que "guía" la reacción para que se forme el producto deseado. Después, la ayuda se puede quitar, dejando el producto puro.

Las oxazolidinonas son un poco caras, pero este método es muy confiable y fácil de usar, por lo que es muy popular en los laboratorios.
Reacción Aldólica de Mukaiyama
La reacción aldólica de Mukaiyama es otro tipo de reacción aldólica que usa éteres enólicos de sililo y es ayudada por "ácidos de Lewis" (sustancias que aceptan electrones). Esta reacción es especialmente útil para ciertos tipos de aldehídos que son difíciles de usar en otros métodos.
Reacciones Aldólicas Organocatalíticas
Un avance más reciente es el uso de "organocatalizadores". Estos son catalizadores (sustancias que aceleran una reacción sin consumirse) que están hechos de pequeñas moléculas orgánicas, no de metales. Un ejemplo famoso es el uso de la prolina, un aminoácido, para catalizar la ciclización de una tricetona.

Esta estrategia es muy poderosa porque permite generar productos enantiopuros sin usar metales de transición, que a veces pueden ser tóxicos o caros. Además, permite realizar reacciones aldólicas cruzadas entre dos aldehídos, lo cual es muy difícil de lograr de otra manera.
Un ejemplo impresionante de esto fue la síntesis de carbohidratos (azúcares) por un grupo de científicos en 2004. Usando organocatalizadores, lograron sintetizar azúcares complejos en solo dos pasos, mientras que los métodos tradicionales requerían muchos más.

Adiciones Aldólicas "Directas"
Normalmente, en una reacción aldólica, primero se forma un enolato, luego reacciona y finalmente se añade un protón. Las "adiciones aldólicas directas" buscan simplificar esto, haciendo que la reacción ocurra en menos pasos. Esto puede hacer que el proceso sea más eficiente y económico para la industria.
Galería de imágenes
-
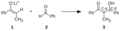
Adición aldólica típica: un enolato de cetona típico 1, actuando como nucleófilo, se adiciona al carbono electrofílico de un aldehído 2, formándose un nuevo enlace carbono-carbono, obteniéndose el producto aldólico 3.
-
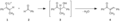
Condensación aldólica típica: un enolato de cetona 1, se adiciona al carbono electrofílico de un aldehído 2, formándose el producto de adición 3. Este producto intermedio pierde una molécula de agua, obteniéndose el producto de condensación 4.
-
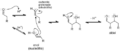
Mecanismo para la reacción aldólica catalizada por ácido de un aldehído consigo mismo.
-
Mecanismo para la deshidratación de un aldol catalizada por ácido.
-
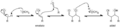
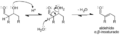
Mecanismo simple para la reacción de aldolización catalizada por base de un aldehído consigo mismo.
-
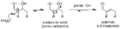
Mecanismo simple para la deshidratación de un producto aldólico.
-
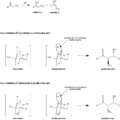
Modelo de Zimmerman-Traxler.
-
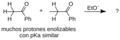
Reacción aldólica hipotética.
-

Cuatro productos posibles de la reacción aldólica.
-
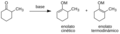
Enolato cinético y termodinámico.
-
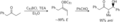
Formación anti del aldol vía un Z-enolato.
-
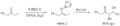
Formación syn del aldol vía un E-enolato.
-
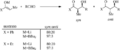
Efecto del ion metálico en el estado de transición.
-

Reacción aldólica con estereocontrol basado en enolato.
-

Modelo general de la reacción aldólica con estereocontrol basado en enolato.
-
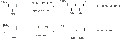
Reacción aldólica con estereocontrol basado en enolato.
-

Modelo general de una reacción aldólica con estereocontrol basado en el carbonilo.
-
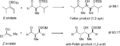
Ejemplos de reacción aldólica con estereocontrol basado en carbonilo.
-

Ejemplos de aplicación del modelo combinado para la estereoinducción.
-
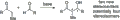
La reacción aldólica crea estereoisómeros.
-

Los cuatro posibles estereoisómeros de la reacción aldólica.
-

Acilación de una oxazolidinona.
-
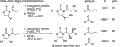
Formación de Z-enolatos usando enolización suave mediada por boro.
-
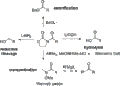
Ruptura de la oxazolidinona quiral de Evans.
-

Reacción aldólica acetato usando un grupo tioéter temporal.
-

Reacción aldólica de Mukaiyama con acetales de sililcetena.
-
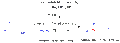
Proceso aldólico vinílico análogo de Mukaiyama.
-

Reacción aldólica con tiazoldinetiona crimina.
-

Mecanismo de reacción de las reacciones aldólicas catalizadas por prolina.
-
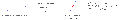
Reacción aldólica cruzada entre dos aldehídos catalizada por organocatalizadores.
-
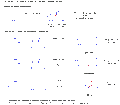
Síntesis de glucosa, manosa y alosa.
-

Adición aldólica directa.
-

Sililación del aducto aldólico.
-
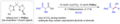
Adición aldólica directa biomimética.












Véase también
 En inglés: Aldol reaction Facts for Kids
En inglés: Aldol reaction Facts for Kids
