
Síndrome de Edwards para niños
Datos para niños Síndrome de Edwards |
||
|---|---|---|
 Cromosoma 18
|
||
| Especialidad | genética médica pediatría |
|
| Síntomas | Cabeza pequeña, mandíbula pequeña, puños cerrados con dedos superpuestos, discapacidad intelectual profunda | |
| Complicaciones | Defectos cardíacos | |
| Causas | Tercera copia del cromosoma 18 (generalmente nueva mutación) | |
| Diagnóstico | Ultrasonidos, amniocentesis | |
El síndrome de Edwards, también conocido como trisomía 18, es una condición genética. Se produce cuando una persona tiene una copia extra del cromosoma 18. Normalmente, tenemos dos copias de cada cromosoma. En este síndrome, hay tres copias del cromosoma 18.
A veces, la copia extra es un cromosoma 18 completo. Otras veces, solo una parte del cromosoma 18 está duplicada. Esto puede ocurrir en todas las células del cuerpo o solo en algunas.
Esta condición fue descrita por primera vez por el científico John H. Edwards en 1960. Sus hallazgos fueron muy importantes para la medicina y la genética.
Contenido
¿Por qué ocurre el Síndrome de Edwards?
El síndrome de Edwards se debe a un error en los cromosomas. Los cromosomas son como paquetes que guardan nuestra información genética.
Errores en la división celular
La causa más común es un error cuando las células se dividen. Esto sucede antes de que se forme el bebé. Normalmente, cuando un óvulo o un espermatozoide se forman, cada uno recibe una sola copia de cada cromosoma. Pero a veces, el cromosoma 18 no se separa correctamente. Así, el óvulo o el espermatozoide terminan con dos copias del cromosoma 18 en lugar de una.
Cuando este óvulo o espermatozoide se une con el otro, el bebé tendrá tres copias del cromosoma 18. Esto se llama trisomía 18. Los científicos siguen investigando por qué ocurren estos errores.
Otras formas del síndrome
En algunos casos, el síndrome de Edwards puede ocurrir de otras maneras:
- Translocación: Un pequeño pedazo del cromosoma 18 se pega a otro cromosoma. Las personas con esta forma suelen tener características menos marcadas.
- Mosaicismo: Algunas células del cuerpo tienen la copia extra del cromosoma 18, mientras que otras células tienen el número normal de cromosomas. Las características del síndrome pueden variar mucho en estos casos.
¿Qué características tiene el Síndrome de Edwards?
Las personas con síndrome de Edwards pueden tener varias características físicas y de salud. Aquí te mostramos algunas de las más comunes:

Características físicas
- En la cara:
* Cabeza más pequeña de lo normal (microcefalia). * Parte de atrás de la cabeza prominente. * Frente estrecha. * Ojos con aberturas pequeñas. * Orejas bajas y con forma diferente. * Mandíbula pequeña (micrognatia).
- En el cuerpo y extremidades:
* Cuello con pliegues de piel. * Esternón (hueso del pecho) corto. * Pies con una forma especial, a veces llamados "pie en mecedora". * Dedos de las manos superpuestos, donde el segundo dedo se cruza sobre el tercero y el quinto sobre el cuarto. * Uñas pequeñas. * Puños cerrados con fuerza.
Desafíos de salud
Las personas con síndrome de Edwards pueden tener desafíos en diferentes partes del cuerpo:
- Corazón: Muchos tienen problemas en el corazón desde el nacimiento.
- Cerebro: Pueden tener algunas diferencias en el desarrollo del cerebro.
- Sistema digestivo: A veces presentan hernias o problemas en los intestinos.
- Riñones y sistema urinario: Pueden tener riñones con formas diferentes o problemas en su funcionamiento.
- Músculos: Pueden tener músculos con menos firmeza de lo normal (hipotonía).
¿Cómo se diagnostica el Síndrome de Edwards?
El diagnóstico del síndrome de Edwards se puede hacer antes o después del nacimiento.
Durante el embarazo
Los médicos pueden sospechar el síndrome durante el embarazo. Esto se hace con ecografías (ultrasonidos). Las ecografías pueden mostrar algunas de las características físicas del síndrome.
Si hay sospechas, se pueden hacer pruebas más específicas. Estas pruebas toman una pequeña muestra de células del bebé o de la placenta. Luego, se analizan los cromosomas para confirmar si hay una copia extra del cromosoma 18.
Después del nacimiento
Después del nacimiento, los médicos pueden identificar el síndrome al observar las características físicas del bebé. También se pueden hacer pruebas genéticas para confirmar el diagnóstico.
¿Qué tan común es el Síndrome de Edwards?
El síndrome de Edwards ocurre en aproximadamente 1 de cada 6000 nacimientos. Es importante saber que el riesgo de tener un bebé con síndrome de Edwards aumenta un poco con la edad de la madre.
Galería de imágenes
-
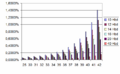
Riesgo de Trisomía 18 (Síndrome de Edwards) en relación con la edad de la madre (eje X) y la gestación (Hbd, marcado por color).

Véase también
 En inglés: Trisomy 18 Facts for Kids
En inglés: Trisomy 18 Facts for Kids
