
Adsorción para niños
La adsorción es un proceso en el que átomos, iones o moléculas de un gas, líquido o sólido disuelto se pegan a una superficie. Imagina que tienes una esponja y la pones en contacto con agua. Si el agua se queda solo en la superficie de la esponja, eso es adsorción. Si el agua entra y llena todos los agujeros de la esponja, eso es absorción. La adsorción es un fenómeno que ocurre solo en la superficie de un material, mientras que la absorción afecta todo el volumen del material. El término sorción se usa para referirse a ambos procesos, y desorción es lo contrario, cuando las moléculas se despegan de la superficie.
La adsorción ocurre debido a la energía que tienen las superficies de los materiales. Los átomos en la superficie de un material no están completamente rodeados por otros átomos de ese mismo material, lo que les permite atraer y sujetar otras moléculas. La forma en que se unen estas moléculas puede ser de dos tipos principales:
- Fisisorción: Las moléculas se unen con fuerzas débiles, como las fuerzas de van der Waals. Es como un imán débil.
- Quimisorción: Las moléculas se unen con enlaces más fuertes, como los enlaces covalentes. Es como un pegamento más resistente.
La adsorción es muy común en la naturaleza y se usa en muchas aplicaciones industriales. Por ejemplo, se utiliza en los catalizadores (sustancias que aceleran reacciones químicas), en el carbón activado para purificar el aire o el agua, y en sistemas para almacenar energía.
La palabra "adsorción" fue creada en 1881 por el científico alemán Heinrich Kayser.
Contenido
¿Cómo se describe la adsorción? Las isotermas

Para entender cómo se pegan los gases o las sustancias disueltas a una superficie, los científicos usan algo llamado isotermas. Una isoterma es un gráfico que muestra cuánta sustancia se ha pegado a un material a una temperatura constante, dependiendo de la presión (si es un gas) o la concentración (si es una sustancia disuelta en un líquido). La cantidad adsorbida casi siempre se mide por la masa del material adsorbente para poder comparar diferentes materiales. Existen muchos modelos de isotermas, pero aquí veremos algunos de los más importantes.
Isoterma de Freundlich
El primer modelo matemático para describir la adsorción fue propuesto por Freundlich y Kuster en 1906. Es una fórmula que se basa en observaciones y se usa para gases:
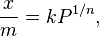
Donde 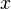 es la masa de la sustancia adsorbida,
es la masa de la sustancia adsorbida, 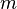 es la masa del material adsorbente,
es la masa del material adsorbente, 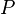 es la presión de la sustancia (o su concentración si es una solución), y
es la presión de la sustancia (o su concentración si es una solución), y 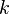 y
y 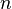 son números que se encuentran experimentalmente para cada material y sustancia a una temperatura dada. Esta fórmula funciona bien para presiones bajas y medias, pero no para presiones muy altas.
son números que se encuentran experimentalmente para cada material y sustancia a una temperatura dada. Esta fórmula funciona bien para presiones bajas y medias, pero no para presiones muy altas.
Isoterma de Langmuir
Irving Langmuir fue el primero en crear un modelo de adsorción con una base científica en 1918. Este modelo se usa para gases que se pegan a superficies sólidas. Es muy popular porque es sencillo y se adapta a muchos datos de adsorción. Se basa en algunas ideas clave:
- Todos los lugares donde se pueden pegar las moléculas en la superficie son iguales, y cada lugar solo puede sujetar una molécula.
- La superficie es uniforme y las moléculas pegadas no interactúan entre sí.
- No hay cambios de estado (como de gas a líquido).
- Cuando la adsorción es máxima, solo se forma una capa de moléculas sobre la superficie.
Aunque estas ideas no siempre son completamente ciertas en la realidad (las superficies pueden tener imperfecciones o las moléculas pueden interactuar), el modelo de Langmuir es un buen punto de partida para entender la adsorción y se usa mucho en el estudio de las reacciones en superficies.
Langmuir propuso que la adsorción ocurre así: una molécula de gas (A) se une a un lugar libre (S) en la superficie para formar AS. En equilibrio, la cantidad de lugares ocupados (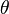 ) se puede calcular con la siguiente fórmula:
) se puede calcular con la siguiente fórmula:
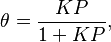
Donde es la presión del gas y 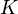 es una constante. Esta fórmula nos ayuda a entender cómo la cantidad de gas adsorbido cambia con la presión.
es una constante. Esta fórmula nos ayuda a entender cómo la cantidad de gas adsorbido cambia con la presión.
Isoterma BET

A veces, las moléculas no solo forman una capa sobre la superficie, sino que se pegan unas sobre otras, formando varias capas. El modelo de Langmuir no puede explicar esto. En 1938, Stephen Brunauer, Paul Emmett y Edward Teller desarrollaron un modelo que sí considera esta posibilidad, llamado la teoría BET. Ellos modificaron la idea de Langmuir para incluir la formación de múltiples capas.
La fórmula BET es más compleja, pero permite calcular la cantidad de sustancia adsorbida cuando se forman varias capas. Una idea importante de este modelo es que el calor necesario para que las moléculas se peguen en las capas superiores es similar al calor necesario para que un gas se convierta en líquido. El modelo BET es muy útil para entender la fisisorción en superficies que no tienen poros muy pequeños.
Isoterma de Kisliuk

En algunos casos, las moléculas de gas que ya están pegadas a una superficie pueden influir en cómo se pegan las nuevas moléculas. Paul Kisliuk estudió este efecto en 1957. Él propuso la teoría del "estado precursor", donde las moléculas de gas primero entran en un estado intermedio en la superficie antes de pegarse definitivamente o volver al gas. La probabilidad de que se peguen depende de si hay otras moléculas ya adsorbidas cerca. Este modelo es más complejo y se usa para situaciones específicas donde las interacciones entre moléculas son importantes.
Entalpía de adsorción
La entalpía de adsorción es la energía que se libera o se absorbe cuando una sustancia se pega a una superficie. Es como el "calor" del proceso. Esta energía nos ayuda a entender qué tan fuerte es la unión entre la sustancia y la superficie.
Explicación a nivel molecular
La adsorción de moléculas en una superficie es un equilibrio entre dos procesos: la adsorción (cuando las moléculas se pegan) y la desorción (cuando se despegan). Si se pegan más moléculas de las que se despegan, la cantidad de sustancia en la superficie aumenta. La velocidad a la que se pegan las moléculas depende de la temperatura, de cómo se mueven las moléculas y de la energía necesaria para que se unan a la superficie. La velocidad a la que se despegan depende de qué tan fuerte estén unidas y de la temperatura.
Materiales adsorbentes

Los materiales que se usan para adsorber se llaman adsorbentes. Suelen tener forma de pequeños gránulos o varillas, con un tamaño de entre 0.25 y 5 milímetros. Para ser buenos adsorbentes, deben ser muy resistentes al desgaste, estables a altas temperaturas y tener muchos poros pequeños. Esto les da una gran superficie para que las moléculas se peguen y permite que los gases se muevan rápidamente a través de ellos.
Los adsorbentes más comunes en la industria se dividen en tres grupos:
- Compuestos con oxígeno: Son materiales que atraen el agua (hidrófilos) y son polares. Ejemplos son el gel de sílice y las zeolitas.
- Compuestos a base de carbono: No atraen el agua (hidrófobos) y no son polares. Incluyen el carbón activado y el grafito.
- Compuestos a base de polímeros: Pueden ser polares o no polares, dependiendo de su composición.
Gel de sílice

El gel de sílice es un material seguro, no tóxico y muy estable. Se usa para secar el aire en procesos industriales (como el aire para producir oxígeno o gas natural) y para eliminar sustancias pesadas del gas natural.
Zeolitas
Las zeolitas son materiales cristalinos que pueden ser naturales o fabricados. Tienen una estructura con poros que se repiten y liberan agua cuando se calientan. Son materiales polares. Se usan para secar el aire, eliminar el dióxido de carbono del gas natural, separar gases y en procesos catalíticos. También existen zeolitas no polares que se usan para fines específicos.
Carbón activado

El carbón activado es un material muy poroso, generalmente en forma de pequeños gránulos o polvo. Es un material no polar y económico. Se fabrica a partir de materiales ricos en carbono como el carbón, la madera o las cáscaras de nuez. El proceso de fabricación tiene dos fases:
- Carbonización: Se calienta el material a más de 400 °C en un ambiente sin oxígeno para eliminar impurezas.
- Activación: El material carbonizado se expone a vapor o dióxido de carbono a alta temperatura. Esto crea una red de poros en el carbón, aumentando enormemente su superficie.
El carbón activado es el adsorbente más usado porque sus propiedades se pueden ajustar para diferentes necesidades. Es excelente para adsorber sustancias orgánicas y no polares, y se usa mucho en el tratamiento de gases y aguas residuales.
Aplicaciones de la adsorción
Adsorción de agua
La adsorción de agua en las superficies es muy importante en la química y la ciencia de materiales. La presencia de agua pegada a las superficies de los sólidos puede influir en cómo se comportan los materiales y en las reacciones químicas. El agua puede pegarse de forma física (se puede quitar secando) o química (las moléculas de agua se rompen y se unen a la superficie).
Calefacción y almacenamiento de energía solar
La adsorción se puede usar para almacenar energía solar. Por ejemplo, las zeolitas sintéticas pueden almacenar calor del sol como energía química. El aire caliente de los paneles solares se hace pasar por un lecho de zeolita, que absorbe la humedad. Cuando se necesita calor (por la noche o en invierno), se hace pasar aire húmedo por la zeolita. Al adsorber la humedad, la zeolita libera calor, que se puede usar para calentar un edificio.
Captura y almacenamiento de carbono
La adsorción se investiga como una forma de capturar el dióxido de carbono de la atmósfera o de las industrias. Materiales como las zeolitas y los MOF (estructuras metal-orgánicas) pueden adsorber CO2. La ventaja es que estos adsorbentes se pueden regenerar (liberar el CO2) usando menos energía que otros métodos.
Adsorción de proteínas
La adsorción de proteínas es muy importante en el campo de los biomateriales (materiales que interactúan con sistemas biológicos). Cuando un biomaterial entra en contacto con fluidos del cuerpo, como la sangre, se cubre rápidamente con una capa de proteínas. Las células del cuerpo no interactúan directamente con el biomaterial, sino con esta capa de proteínas. Esta capa de proteínas "traduce" las propiedades del biomaterial a un "lenguaje biológico" que las células pueden entender, influyendo en cómo las células se pegan, crecen y se desarrollan.
Enfriadores de adsorción

Los enfriadores de adsorción usan el calor (por ejemplo, calor residual de industrias o energía solar) para producir frío. Funcionan combinando un material adsorbente con un refrigerante. El material adsorbente (como zeolita o carbón activado) adsorbe el refrigerante. Cuando se calienta, el material libera el vapor refrigerante, que luego se enfría y se vuelve líquido, produciendo un efecto de enfriamiento. Después, el vapor refrigerante vuelve a ser adsorbido por el material. Estos enfriadores son bastante silenciosos porque no necesitan un compresor.
Adsorción en virus
La adsorción es el primer paso en el ciclo de vida de un virus. Es cuando el virus se pega a la superficie de una célula. Después de pegarse, el virus entra en la célula, libera su material genético, lo usa para hacer más copias de sí mismo y finalmente libera nuevos virus.
En la cultura popular
El juego de Tetris se ha usado como un ejemplo para estudiar la adsorción. Los científicos han utilizado los bloques de Tetris como si fueran moléculas de formas complejas que se pegan a una superficie plana. Esto les ayuda a entender cómo se comportan las nanopartículas a nivel termodinámico.
Galería de imágenes
-

El modelo de adsorción multicapa de Brunauer, Emmett y Teller es una distribución aleatoria de moléculas en la superficie del material.

Véase también
 En inglés: Adsorption Facts for Kids
En inglés: Adsorption Facts for Kids
