
Reacción de Diels-Alder para niños
La reacción Diels-Alder es una forma especial de unir dos moléculas para crear un anillo de seis átomos de carbono. Es una reacción pericíclica de cicloadición, lo que significa que ocurre en un solo paso y forma un anillo. En esta reacción, un dieno (una molécula con dos enlaces dobles conectados por un enlace sencillo) se une a un dienófilo (una molécula con un enlace doble o triple).
Al mismo tiempo que se forman dos nuevos enlaces carbono-carbono, la reacción Diels-Alder es muy útil para construir anillos de seis miembros. Permite controlar muy bien cómo se unen las moléculas y la forma tridimensional del producto. Por eso, es una herramienta poderosa en la química para crear moléculas complejas, como las que se encuentran en la naturaleza o nuevos materiales.
Esta reacción también se puede usar con átomos diferentes al carbono, como el oxígeno o el nitrógeno, para formar otros tipos de anillos. A esto se le llama reacción hetero-Diels-Alder.
_red.svg)
Contenido
- Historia de la Reacción Diels-Alder
- El Dienófilo: El Compañero de Reacción
- El Dieno: La Molécula con Dos Enlaces Dobles
- Cómo Funciona la Reacción: El Mecanismo
- Regioselectividad: ¿Dónde se unen los átomos?
- Estereoselectividad y Estereospecificidad: La Forma 3D
- Reacción de Diels-Alder Intramolecular
- Reacción Reversa: Retro-Diels-Alder
- Variaciones de la Reacción Diels-Alder
- Aplicaciones en la Síntesis Química
- Reacciones de Diels-Alder Biológicas
- Galería de imágenes
- Véase también
Historia de la Reacción Diels-Alder
¿Quiénes descubrieron la reacción Diels-Alder?
Los químicos Otto Paul Hermann Diels y Kurt Alder fueron los primeros en describir esta reacción en 1928. Por su importante trabajo, recibieron el Premio Nobel de Química en 1950.
El primer ejemplo que describieron fue la reacción entre el 1,3-butadieno y el eteno. También mostraron la reacción entre el ciclopentadieno y la quinona.

Diels y Alder publicaron sus descubrimientos en una serie de 28 artículos entre 1928 y 1937.
El Dienófilo: El Compañero de Reacción
¿Qué es un dienófilo y cómo reacciona?
El dienófilo es la molécula que tiene un enlace doble (o triple) y que se une al dieno. La reacción Diels-Alder ocurre más rápido si el dienófilo tiene un grupo que atrae electrones cerca de su enlace doble.
Por ejemplo, el etileno reacciona lentamente. Pero moléculas como el propenal, el propenoato de etilo o el anhídrido maleico reaccionan mucho más rápido porque tienen estos grupos que atraen electrones.
El Dieno: La Molécula con Dos Enlaces Dobles
¿Cómo debe ser un dieno para reaccionar?
El dieno es la molécula con dos enlaces dobles. Para que la reacción Diels-Alder funcione, el dieno debe tener una forma específica llamada conformación "s-cis". Esto significa que los dos enlaces dobles deben estar del mismo lado del enlace sencillo que los une.
Solo en esta forma "s-cis" los extremos del dieno están lo suficientemente cerca para unirse al dienófilo. Si el dieno está en la forma "s-trans", los extremos están demasiado lejos.

Para que un dieno sea muy reactivo, debe tener grupos que donen electrones. Estos grupos hacen que el dieno sea más "rico" en electrones, lo que facilita la reacción.
Los dienos pueden tener muchas estructuras diferentes, pueden ser de cadena abierta o formar parte de un anillo. Algunos dienos, como el furano y el pirrol, también pueden participar en estas reacciones.

Cómo Funciona la Reacción: El Mecanismo
¿Cómo se unen las moléculas en la reacción Diels-Alder?
La reacción Diels-Alder es un ejemplo de una reacción "concertada". Esto significa que todos los enlaces se rompen y se forman al mismo tiempo, en un solo paso, sin que se creen moléculas intermedias.
Se cree que la reacción ocurre porque los electrones de las moléculas se alinean de una manera especial. Los científicos Woodward y Hoffmann desarrollaron unas reglas que explican por qué esta reacción ocurre tan fácilmente.
La reacción se inicia con calor y no necesita luz. Es interesante que algunas reacciones Diels-Alder son mucho más rápidas en solventes polares, como el agua. Por ejemplo, la reacción del ciclopentadieno y la butenona es 700 veces más rápida en agua que en otros solventes. Esto puede deberse a que las moléculas se agrupan mejor en agua o a que el agua ayuda a estabilizar el estado de transición.
La forma tridimensional de las moléculas iniciales (dieno y dienófilo) determina la forma tridimensional del producto final.
Regioselectividad: ¿Dónde se unen los átomos?
¿Cómo se predice la unión de los átomos en la reacción?
La regioselectividad se refiere a qué átomos específicos del dieno se unen a qué átomos específicos del dienófilo. En general, la reacción Diels-Alder sigue una regla llamada "orto-para", que ayuda a predecir cómo se unirán los sustituyentes (los grupos de átomos unidos a las moléculas principales).
Por ejemplo, si el dieno tiene un grupo que dona electrones en una posición, y el dienófilo tiene un grupo que atrae electrones en otra, la reacción tenderá a formar un producto donde estos grupos estén en posiciones específicas, similares a las posiciones "orto" y "para" en los anillos de benceno.

Estereoselectividad y Estereospecificidad: La Forma 3D
¿Cómo se mantiene la forma de las moléculas en la reacción?
Las reacciones Diels-Alder son "estereoespecíficas". Esto significa que la información sobre la forma tridimensional de las moléculas iniciales se mantiene en el producto. Si los grupos estaban en una posición "cis" (del mismo lado) en el dienófilo, seguirán estando "cis" en el nuevo anillo. Lo mismo ocurre con los grupos en posición "trans" (lados opuestos).

Cuando se forman nuevos centros con una forma tridimensional específica, hay dos posibles resultados. Esto se llama "estereoselectividad". En la reacción Diels-Alder, a menudo se prefiere un tipo de orientación llamado "endo". Esto se conoce como la regla endo de Alder. En el estado de transición "endo", los grupos importantes del dienófilo se orientan hacia el dieno.

La explicación más aceptada para esta preferencia "endo" es una interacción favorable entre los electrones de las moléculas, aunque otros factores como las atracciones entre las moléculas también pueden influir. A veces, si los grupos son muy grandes, los efectos de espacio pueden hacer que se prefiera la orientación "exo" en lugar de la "endo".
Reacción de Diels-Alder Intramolecular
¿Qué pasa si el dieno y el dienófilo están en la misma molécula?
Una reacción intramolecular de Diels-Alder ocurre cuando el dieno y el dienófilo son parte de la misma molécula. La reacción forma la misma estructura de anillo de seis miembros, pero como parte de un sistema de anillos más complejo.
Un ejemplo de esto fue en la síntesis del ácido giberélico, una sustancia importante en las plantas.

Reacción Reversa: Retro-Diels-Alder
¿Puede la reacción Diels-Alder ir hacia atrás?
Sí, la reacción retro-Diels-Alder (rDA) es lo opuesto a la reacción Diels-Alder. En esta reacción, el anillo de seis miembros se rompe para formar de nuevo el dieno y el dienófilo originales.

Esta reacción inversa suele ocurrir a altas temperaturas. Por ejemplo, el ciclohexeno puede romperse en 1,3-butadieno y etileno a temperaturas elevadas.
Variaciones de la Reacción Diels-Alder
¿Existen diferentes tipos de reacciones Diels-Alder?
Hay varias variaciones de la reacción Diels-Alder:
- Reacciones Deshidro-Diels-Alder (DDA): Estas reacciones involucran moléculas con enlaces triples y pueden generar anillos de benceno.
- Catálisis con Ácidos de Lewis: Algunas sustancias llamadas ácidos de Lewis (como ZnCl2 o BF3) pueden acelerar mucho las reacciones Diels-Alder y mejorar la forma en que se unen los átomos.
- Hetero-Diels-Alder: Como se mencionó antes, esta variación ocurre cuando algunos de los átomos en el anillo formado no son carbono, sino otros elementos como el oxígeno o el nitrógeno.
* Oxo-Diels-Alder: Es una variación donde un dieno reacciona con un aldehído (que actúa como dienófilo) para formar un anillo que contiene oxígeno. * Aza-Diels-Alder: En esta reacción, las iminas (moléculas con un enlace doble carbono-nitrógeno) reaccionan con dienos para formar anillos que contienen nitrógeno.

Aplicaciones en la Síntesis Química
¿Para qué se usa la reacción Diels-Alder?
La reacción Diels-Alder es muy importante en la química para construir moléculas complejas.
- El anhídrido maleico es un compuesto clásico que se usa en la reacción Diels-Alder. Se ha utilizado para crear muchos pesticidas y medicamentos.

- El químico R. B. Woodward usó la reacción Diels-Alder en la síntesis de corticoesteroides y colesterol, que son moléculas muy importantes en el cuerpo.

- E. J. Corey la usó para crear prostaglandinas, que son sustancias que regulan muchas funciones en el cuerpo.

- Otros científicos como Samuel J. Danishefsky, Paul Wender y Stephen F. Martin también han usado esta reacción para sintetizar moléculas complejas como el prefenato disódico o la reserpina.
- La reacción Diels-Alder también se ha utilizado para crear el núcleo de algunos antibióticos, como la tetraciclina.
Reacciones de Diels-Alder Biológicas
¿Ocurre la reacción Diels-Alder en la naturaleza?
Sí, se ha descubierto que la reacción Diels-Alder también ocurre en sistemas biológicos, es decir, dentro de los seres vivos, catalizada por enzimas.
Por ejemplo, la enzima macrofomato sintasa utiliza una reacción Diels-Alder para convertir una molécula en ácido macrofómico. Esto sugiere que las reacciones Diels-Alder pueden ser una forma en que la naturaleza construye moléculas complejas.

También se ha postulado que la formación de algunas moléculas como la absintina (que se encuentra en la planta de ajenjo) podría involucrar una reacción de Diels-Alder natural.

Galería de imágenes
-
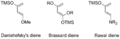
Forma general de los dienos de Danishefsky, Brassard y Rawal.
-
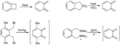
Generación in situ de o-quinodimetanos.
-
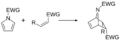
Reacción de Diels-Alder con pirrol.
-
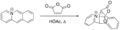
Ejemplo de cicloadición de Bradsher.
-

Diagrama de orbitales moleculares frontera (FMO) en la reacción Diels-Alder.
-
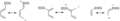
Estructuras de resonancia de dienos y dienófilos.
-
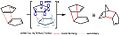
Estado de transición endo en la dimerización del ciclopentadieno.
-

Reacción Deshidro-Diels-Alder.
-
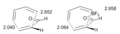
Geometrías del estado de transición de la reacción de Diels-Alder.
-
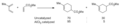
Regioselectividad de una reacción Diels-Alder con y sin catálisis.
-
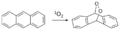
Reacción de Diels-Alder entre antraceno y dioxígeno singlete.
-
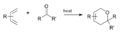
Esquema general de la reacción oxo-Diels-Alder.
-
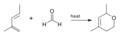
Reacción oxo-Diels-Alder reportada por Gresham en 1949.
-

Reacción Aza-Diels-Alder con bencilamina.
-

Reacción Aza-Diels-Alder enantioselectiva catalizada por S-prolina.
-
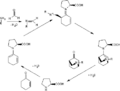
Ciclo catalítico para la reacción Aza-Diels-Alder enantioselectiva catalizada por S-prolina.
-
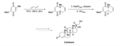
Diels-Alder en la síntesis total de cortisona por R. B. Woodward.
-

Síntesis de prefenato disódico por Danishefsky.
-
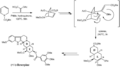
Uso de Diels-Alder en la síntesis de reserpina por Paul Wender.
-
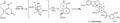
Uso de Diels-Alder en la síntesis de reserpina por Stephen F. Martin.
-
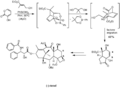
Uso de Diels-Alder en la síntesis de taxol por K. C. Nicolaou.
-
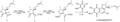
Síntesis de (-) - furaquinocina C por Amos Smith.
-

Síntesis de tabersonina por Viresh Rawal y Sergey Kozmin.
-
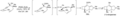
Síntesis enantioselectiva de (+) - esterpureno por William Okamura y Richard Gibbs.
-
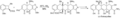
Síntesis de (-) - tetraciclina por Andrew Myers.
-
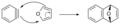
Cicloadición [4+2] entre bencino y furano.
-
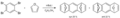
Reacción de diarinos con furano.
-
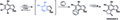
Síntesis de herbindole A por Buszek y colaboradores.
-
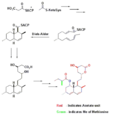
Formación in vitro de una lactona tricétida.






Véase también
 En inglés: Diels–Alder reaction Facts for Kids
En inglés: Diels–Alder reaction Facts for Kids
